Biography
Interests
Dr. Masoumi, M.1,2*, Dr. Amirkanian, F.1, Dr. Tabaraei, R.1, Dr. Salevati Pour, A.1 & Dr. Mullabashi, T.1
1Qom University of Medical Sciences (QUMS), Shahid Beheshti Hospital, Qom, Iran
2Rheumatology Research Center (RRC), Tehran University of Medical Science, Shariati Hospital, Tehran, Iran
*Correspondence to: Dr. Masoumi, M., Qom University of Medical Sciences (QUMS) and Rheumatology Research Center (RRC), Iran.
Copyright © 2018 Dr. Masoumi, M., et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Introduction
Kikuchi-Fujimoto disease (KFD) or histiocytic necrotizing lymphadenitis is a rare, benign, generally self-limiting disease first described in 1972. KFD generally affects Asian women between the ages of 20 and 35 years old. It frequently manifests as an acute or subacute form with painful posterior cervical lymphadenopathy and systemic symptoms like fever, fatigue, and headache [1-4]. More rarely, patients with KFD can also present with leukopenia, hepatosplenomegaly and complaints of fatigue, rash, arthralgia and weight loss [5]. The diagnosis of KFD is based on histopathological findings of lymph node. Although, it is important to consider differential diagnosis in order to exclude more serious conditions such as lymphoma, infections and autoimmune disorders. Fortunately, the disease has a good prognosis without treatment. Nonetheless, if necessary, we can use analgesics and nonsteroid anti-inflammatory drugs for symptomatic treatment [6,7]. Here, we describe a case of a young Iranian female presented with fever and cervical lymph node enlargement associated with night sweats and weight loss who was finally diagnosed as KFD by histological examination of the affected lymph nodes.
Case Presentation
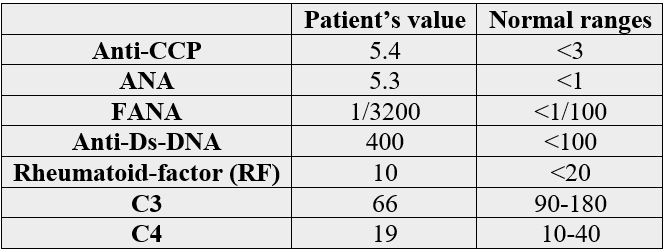
A 29-year-old female patient was admitted to our Internal Medicine Unit on January 2018 with a threemonth history of low grade fever, fatigue, malaise, night sweats, gradual loss of appetite, and 5 kilograms weight loss. She complained chiefly of trouble in walking, a tender lymphadenopathy and a high grade fever that were severed since a week before. Her clinical history was unremarkable and she had no past history of Tuberculosis and contact with it. On admission her heart rate was 88bpm, blood pressure was 120/75mmHg, respiratory rate was 16 breaths/minute, and temperature was 39.1°C. Physical examination revealed asymmetrical additive poly- arthritis involving left hip joint, both knees, left shoulder joint and right metacarpo- pharyngeal and wrist joints. We also detected erythematous lesions on the anterior of chest. Physical examination also revealed bilateral tender, fixed and painful lymphadenopathies that were 3×3cm on the left side and 2×1 - 2×1.5cm on the right side of posterior Jugular triangle palpated in the supraclavicular area. Thus two non-tender, fixed and enlarged lymph nodes were detected in the auxiliary and genitalia. There were no hepatosplenomegaly or other clinically appreciable lymphadenopathy elsewhere. Examination of respiratory and other systems was normal. Laboratory analysis showed slight increase in C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR) values that were, respectively, of 14.5mg/L (n.v.˂6) and 69mm/h. Thyroid hormones and neoplastic markers were in the normal range. HIV ab, cytomegalovirus ab IgM, herpes virus ab IgM, Epstein-Barr virus ab IgM, Brucellosis serology, and Toxoplasma ab igM were all negative. Chest X-ray was un-remarkable except for bilateral segmental opacities in the inferior lobes and bilateral lymphadenopathies in the posterior Jugular triangle. Breast Ultrasonography revealed multiple bilateral enlarged lymphadenopathies with thick cortices. Abdominal CT-scan revealed hepatomegaly and mild ascitic fluid in the pelvic area, plus several small cystic hypo-dense lesions in left ovary. Further rheumatologist’s laboratory tests were ordered and they were positive for Anti-CCP, antinuclear antibody and anti-ds-DNA. The results are in the table below:
Because of a dilemma in clinical diagnosis and the persistence of symptoms, an excision biopsy of the largest supraclavicular lymph node was performed. Sections showed partially effaced lymph node structure with focal well-defined paracortical necrotizing lesion, composed of multiple karyorrhectic debris, scattered fibrin deposition and collections of histiocytes and lymphocytes. Plasma cells, neutrophils and eosinophils were very scant.
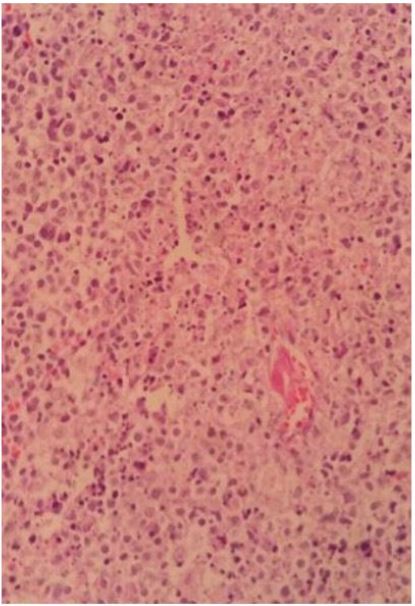
Image show well-defined necrotic foci with nuclear debris surrounded by some mononuclear cells.
Histological examination showed a reactive lymphadenitis and intrafollicular hyperplasia, with the presence of small to large CD5+/-, CD4-/+, CD3+, CD8+/- T lymphocytes, in addition to numerous histiocytes expressing CD68PGM1/MPO and abundant apoptotic nuclear debris. No polymorphonuclear neutrophils and no caseous necrosis were observed. On the basis of these morphological and immunophenotype findings, the diagnosis of Kikuchi-Fujimoto disease was made. Due to positive tests of Systemic Lupus Erythematosus (SLE), an accompanying diagnosis of SLE was made too. The patient was treated symptomatically with Prednisolone, and Hydroxychlorquine and in two-month follow-up, malaise, fatigue, and appetite were improved; she denied fever and night sweats, and ultrasonography showed a decrease in the sizes of supraclavicular, axillary, and genitalia lymph nodes.
Discussion
KFD is a benign self-limiting extremely rare disease known to have a worldwide distribution with a higher prevalence among Japanese and other Asian young (mean, 29 years old) female (male-to-female ratio, 1:4) individuals [1,8].
Particularly, to our best knowledge, only 18 Iranian cases of KFD have been published in the last 20 years witch all patients being younger than 40 years old except two (one 41, and another 51), and two patients accompanying systemic lupus erythematosus (SLE) [9-21]. Patients with KFD usually present with nonspecific signs and symptoms mainly characterized by enlarged, tender unilateral posterior cervical lymph nodes and fever. Less common symptoms are weakness, loss of appetite, night sweats, weight loss, joint pain, or cutaneous manifestations [1-4]. The etiopathogenesis of KFD is still unknown. Certain causative organisms have been proposed. These include Epstein-Barr virus, human T cell leukemia virus type 1, human herpesvirus type 6, B19 parvovirus, cytomegalovirus, Brucella, Yersinia enterocolitica, and parainfluenza virus [22]. It is suggested that the disease is associated with systemic lupus erythematosus (SLE), due to their unclear pathogenic links [23,24]. Nevertheless, during the past years, some theories were developed. One theory involves molecular mimicry, in which infectious agents that closely resemble a host peptide affect the ability of T cells to detect self from nonself [25,26]. Another, suggest that apoptotic cells can be the source of the autoantigens of SLE [27]. Morphological differential diagnosis of necrotizing lymphadenitis including lymphoma, both Hodgkin and non-Hodgkin, autoimmune related lymphadenopathy particularly SLE, infectious diseases such as tuberculosis, infectious mononucleosis and toxoplasmosis. The exclusion of these differential diagnosis is very important because the management of these disease are different.
Differentiating kikuchi disease from lymphoma is crucial due to different treatment and prognosis. Although Hodgkin lymphoma may have necrosis and histiocytes infiltration but in our case, there is no large Reed- Stenberg cells or other variants of Hodgkin cells, no eosinophils or neutrophils infiltration, so the Hodgkin lymphoma excluded.
Also we didn’t have any immunoblast proliferation or collections of plasmacytoid dendritic cells and the cells population is more polymorphic without atypia and increased mitoses. The lymph node architecture is partially preserved and the sinus component is patent. So, no features of non-Hodgkin lymphoma present.
SLE and kikuchi disease both occur in young woman and the SLE lymphadenitis is one of the main challenging diagnosis for kikuchi disease [1]. Hematoxylin bodies are present in SLE lymphadenitis along with presence of follicular hyperplasia, many plasma cells and more neutrophils.
For differentiating kikuchi from infectious etiologies, absence of neutrophils, absence of granulomatous inflammation and viral cytopathic effect, presence of more mononuclear cells and negative staining for Acid fast bacilli was helpful.
A differential diagnosis of malignant lymphoma and infectious lymphadenitis (EBV, Herpes and Toxoplasma) was also considered. Final diagnostic key is lymph node biopsy and histopathological study [22,28]. Since the disease has an unknown etiology, any specific therapy is unavailable. It is usually self-limited; however, Corticosteroid therapy is used only for the most severe cases [1].
Conclusion
In conclusion, despite its benign and self-limited nature, the authors highlight the importance of an accurate diagnosis and also the importance of considering associated conditions like SLE which may need an accompanying therapy. Recalling our case report, the authors also emphasize the fact that patients with Kikuchi-Fujimoto disease may present non-typical cutaneous lesions, which are important in differential diagnoses.
Bibliography


Hi!
We're here to answer your questions!
Send us a message via Whatsapp, and we'll reply the moment we're available!