Biography
Interests
Shimon Shatzmiller*, Inbal Lapidot & Galina Zats-Mordechai
Department of Chemical Sciences, Ariel University, Israel
*Correspondence to: Dr. Shimon Shatzmiller, Department of Chemical Sciences, Ariel University, Israel.
Copyright © 2018 Dr. Shimon Shatzmiller, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
It is 20 years that the acceptance of the Amyloid hypothesis [1] by many researchers that worked very intensively on that basis to find remedy for neurodegenerative diseases. The current state of the art is: although this gave abundant opportunities, only 5 drugs, which do not cure the diseases, are approved by the FDA and those are for palliative therapies. There are new perspectives that combine many of the Amyloid diseases under one roof. The relations between Diabetes II and Alzheimer’s (type 3 diabetes [2]). That approach might open new avenues for drug developments. The paper discuses some older and modern approaches to base drug development research.
Introduction
Alzheimer’s disease (AD) is the most generic form of dementia in the elderly and its prevalence is set to
increase rapidly in coming decades. However, there are yet no available drugs that can halt or even stabilize
disease progression. One of the main pathological features of AD is the presence in the brain of senile
plaques mainly composed of aggregated β amyloid (Aβ), a derivative of the longer amyloid precursor protein
(APP).
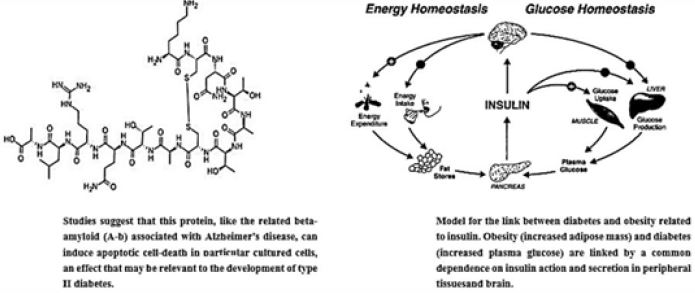
The amyloid hypothesis proposes that the accumulation of Aβ within neural tissue is the initial event that triggers the disease. The Amyloid hypothesis [3] suggests Amyloid-β (Aβ) as main peptides produced in the brain by digestion of the amyloid precursor peptide (APP) by β, and secretase enzymes [4,5]. β amyloid aggregation inhibitors can be small molecules as candidate drugs for therapy of Alzheimer’s disease [6a,b]. Also Amylin is a 37-amino-acid polypeptide [7] which is co-secreted with insulin [8]. After its identification in 1986 [9], there has been a rapid expansion of knowledge relating to amylin, which has already been translated into clinical practice. Amylin has been established as a glucoregulatory, neuroendocrine hormone in animals [10]; however, its role in human physiology is less well defined. Pramlintide, an amylin analogue, has been developed, and is now used in the treatment of diabetes. It is a pancreatic enzyme characteristic to Diabetes Mellitus type II (DM), the “Islet peptide”, is playing a key role in the development of neurodegenerative disorders [5b,11]. The Aβ are antimicrobial enzymes. Aβ and Amylin are both antimicrobial peptides. Also, the Amylin Protein of Diabetes Mellitus is an Antimicrobial Peptide [12].
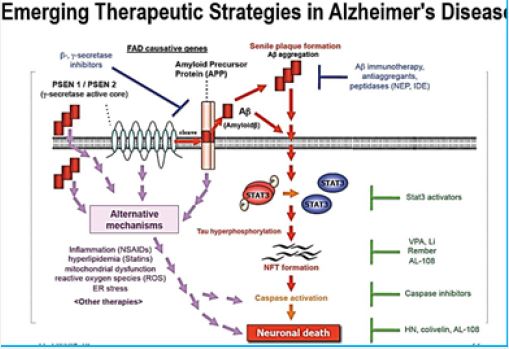
Conformational disease is a general term often used to identify a number of disorders that are characterized by aggregation and deposition of specific proteins. Although the physiological roles of some of these aggregation-prone proteins have yet to be fully elucidated [13], it is possible to distinguish several different pathologies depending on the aggregation of a specific protein, i.e. b-amyloid for Alzheimer’s disease [14], a-synuclein for Parkinson’s disease, etc. In these diseases, the proteins in question are known to convert from their native functional state into highly organized fibrillar aggregates. In the case of amylin [15,16], the observation of amyloid deposits in islets Langerhans of individuals with type 2 diabetes was for a long time considered not important, probably because while researchers studying other pathologies were looking for a protein to blame that could be responsible for the possible pathogenic mechanism, the research field of type 2 diabetes was already well established and mainly directed towardsthe study of insulin resistance [17]. possible pathogenic mechanism, the research field of type 2 diabetes was already well established and mainly directed towards the study of insulin resistance [18].
Currently, only palliative therapies [19] for the AD are available. Acetylcholine inhibitors [20] like Donepezil, Galantamine, and the NMDA receptor [21] antagonist Memantine have been approved for clinical use as a treatment for cognitive symptoms. The five drugs are currently available: four cholinesterase inhibitors and a N-methyl-D-aspartate receptor antagonist.
However, these are only effective for a limited amount of time, between 6 to 12 months, and for half of the patients affected with milder forms of Alzheimer’s. The current FDA approved drugs for the AD do not cure the disease, they are targeted to aid in the misery of the terminal disease by soothing their pains and helplessness, as agents that provide better function of the synaptic processes. Some believe that the inhibitors preventing A aggregation are a new doorway toward new drugs to cure Alzheimer’s disease [22].
Small molecule probes of protein aggregation [23] are a strategy accepted in neurodegenerative disease research for more than two decades. During that time the complex and obscure situation has become clearer. However, many protein misfolding are contributing to neurosecretion [10,24] and are finally appearing as different, but related diseases.
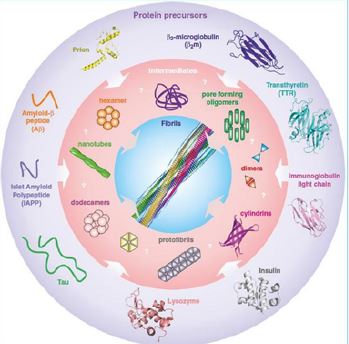
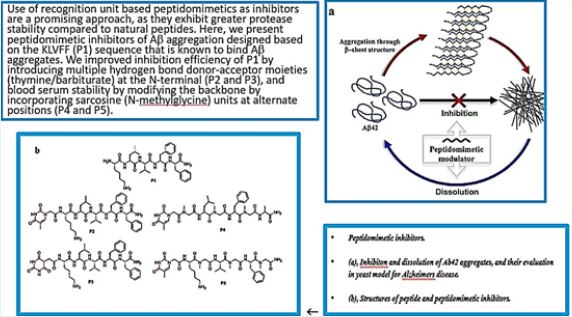
Sketches or structures, where available, of amyloid precursors and peptides associated with various human pathologies. PDB IDs: b2-microglobulin D76N, 4fxl, transthyretin, 3cn4, immunoglobulin light chain, 1bre , insulin, 1zeh , lysozyme, 5fhw, human prion: 1i4m . These unrelated amyloid sequences assemble into highly organized cross-b amyloid fibrils (center) via an array of oligomers that, thus far, have largely eluded structural characterization. A number of possible oligomeric structures have been proposed, including dimers, domain swapped dimers , hexamers and dodecamers , cylindering , nanotubes and pore forming oligomers. The structure of (insulin) fibrils shown in the center is taken from, with permission. Credit ref. 10
-Helix and Β-Sheet Breakers and Peptidomimetics
Synthetic peptide derivatives, termed ‘β-sheet breakers’ [10], have been developed that are able to inhibit
amyloid formation by binding to monomeric precursors, or by preventing fibril elongation by blocking fibril
ends. Substitution of key residues in synthetic peptides corresponding to the amyloid core regions with
prolines, or incorporating N-methyl modified amino acids [25], prevents hydrogen bond formation crucial
to the cross-β structure. Clever use of these strategies within cyclic peptides has been used to create amyloid
inhibitors and has enabled oligomeric intermediates to be isolated and characterized [26].
Foldamers are a very prominent class of α-helix mimetic peptides. They are composed of β-amino acid α/β- amino acid oligomers), or N-substituted glycine residues (peptoids). Such foldamers have been shown to inhibit the proteolytic activity of γ-secretase, an enzyme that is involved in the processing of amyloid-β (Aβ) in Alzheimer’s disease, by blocking the initial substrate binding site of γ-secretase [27, 32].
The “Amyloid peptides” tend to aggregate in the brain to form the plaques and tangles that destroy the neurons in the brain [28] byways that end in apoptosis of the nerve cells, probably by the destruction of the synapses [29].
As illustrated above, more and more evidence accumulates showing that there is a connection between neurosecretion and diabetes. Reports in the literature point to the role of diabetes peptides, both Insulin, Amylin are active in the brain. They have a role in the development of neurological symptoms [30]. A fragment of Amylin (islet peptide) is a trigger of neurodegeneration leading to Alzheimer’s and Parkinson’s diseases. However, on this basis, only a few attempts to suggest peptide-based research for curing and diagnosing the fatal diseases are published [31].
The Antimicrobial peptides: AA-peptides are enzymatically stable, display excellent antimicrobial activity and show promise in many other biological applications such as cancer,
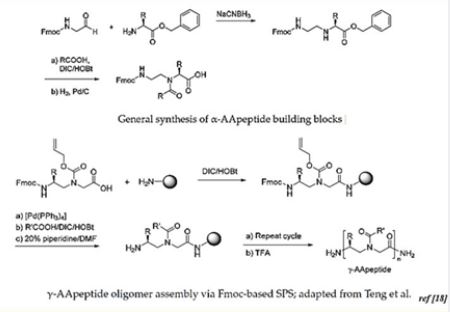
Alzheimer’s disease and biomaterial science [32] AApeptides activity and show promise in many other biological applications such as cancer, Alzheimer´s disease and biomaterial science. [33], the class of peptides in which Aβ and Amylin could be classified, are a currently investigated class of potential drug candidates. Both forms of DM contribute to accelerated cerebral atrophy [34] and to the presence of heightened white matter abnormalities. Rational design of peptide mimics that prevent aggregation of Aβ is shown below [13]:
The combination of established experimental methods with multi-scale state-of-the-art computational methods have shown that castalgine, vescalagin, SEN304 and inh3 are promising molecules to prevent the amyloid- beta peptide aggregation, and to reduce the cellular toxicity of the peptide [35].
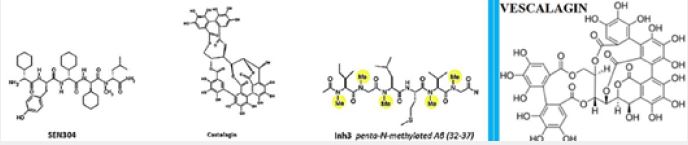
The pioneering work of Ehud Gazit [36], Susanne Aileen Funke and Dieter Willbold [6], the are seeking for peptides that can cross the BBB and interfere with the aggregation of Aβ in the living brain has opened a new avenue for AMP and their surrogates as potential agents for neurodegenerative diseases.
The significance of biological activity is determining factor in search of new drug and diagnostics for neurodegeneration. However, The biological testing can be carried out on many living organisms from worms and fly to mice and more complex organisms. Such tests are cumbersome and expensive. The fact that antimicrobial agents like peptide [37]and their surrogates are potential candidates can have carried us in a simple Minimum Inhibitory Concentrations (MIC) experiments experiments and the indication for toxicity in a hemolysis determination of human red blood cells. The preliminary antimicrobial test approach may accelerate the pace in which candidates then tested for an AD for example.
Peptides and Peptide Surrogates in Amyloid Diseases Research
Despite great diversity in the primary amino acid sequence of proteins, the oligomers of different misfolded
[38] amyloidogenic proteins (hyper phosphorylated tau [39], amyloid-synuclein) elicit toxicity by common
mechanisms. This toxicity involves membrane perturbations, calcium dysregulation, the accumulation of
reactive oxygen species, endoplasmic reticulum (ER)-stress, impairment of the ubiquitin-proteasome system,
and the activation of apoptotic signaling pathways. Indeed, it is hypothesized that the initiation of NDDs is
a result of ER-stress, damaged mitochondria, and loss of Ca2+- and protein homeostasis.
It is reported that γ-peptide-based small-molecule ligands that inhibit Aβ aggregation. (GRKKRRQRRRPQ) was able to bind to HIV TAR RNA and BIV TAR RNA with
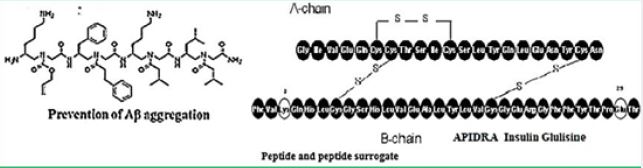
Comparable selectivity and specificity to that of the Tat peptide. Furthermore, -AApeptide mimics of RGD peptides (including 64Cu-labelled compounds), and of fMLF (a selective formyl peptide receptor agonist) have been reported. Also, -AApeptides have been shown to disrupt A aggregation in Alzheimer’s disease, and to have potential applications in biomaterial science.
The classes of antimicrobial peptidomimetics are of interest owing to the convenient synthesis of the corresponding building blocks and their assembly by using standard solid-phase procedures [40].
Dihydropyridine (DHP) Based Agents [41]
We prepared 1,4-Dihydropyridine (DHP) based peptide mimics and had shown that they are antibacterial
agents [42].
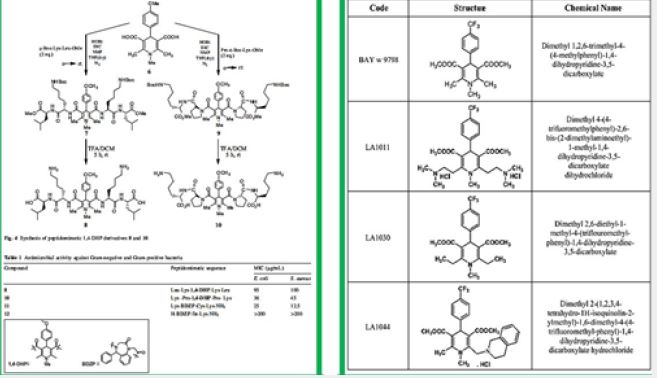
Similar DHP derivatives were shown to have a neuroprotective effect in a transgenic mouse model of Alzheimer’s disease [43].
It was also reported that Selective Antihypertensive Dihydropyridines Lower Aβ Accumulation by Targeting both the Production and the Clearance of Aβ across the Blood-Brain Barrier. Several large populationbased or clinical trial studies have suggested that certain dihydropyridine (DHP) L-type calcium channel blockers (CCBs) used for the treatment of hypertension may confer protection against the development of Alzheimer disease (AD) [44].
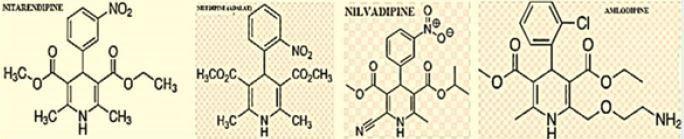
Calcium channel blockers and dementia as reported in the last years by researchers. It became established that In Alzheimer’s disease, enhanced calcium load might be brought about by extracellular accumulation of amyloid-β. Recent studies suggest that soluble forms facilitate influx through calcium-conducting ion channels in the plasma membrane, leading to excitotoxic neurodegeneration.It is based on the “Calcium hypothesis of dementia.”
Among the antihypertensive DHPs tested [45], a few, including nilvadipine, nitrendipine and amlodipine inhibited Aβ production in vitro [28], whereas others had no effect or raised Aβ levels. In vivo, nilvadipine and nitrendipine acutely reduced brain Aβ levels in a transgenic mouse model of an AD and improved Aβ clearance across the blood-brain barrier (BBB), whereas amlodipine and nifedipine were ineffective showings that the Aβ-lowering activity of the DHPs is independent of their antihypertensive activity.
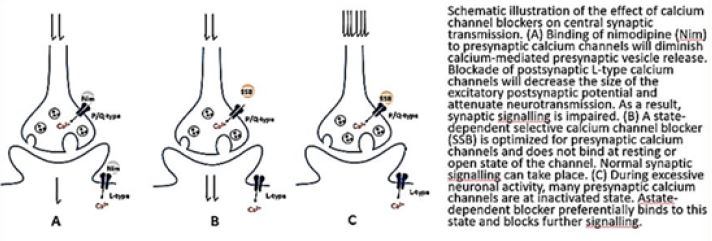
Disruptions of normal calcium (Ca 2+ ) homeostasis in human aging processes, are well documented [27]. The existence of at least four distinct types of Ca 2+ channels, L-, T-, P-, and N-, have been reported in the central nervous system. Binding sites for clinically useful dihydropyridines (DHP) and phenylalkylamines (PA) [46]are located on the L-type Ca 2+ channels. In the present study, DHPbinding parameters were determined in various brain areas obtained at autopsy from Alzheimer’s (AD), Parkinson’s (PD), Huntington’s (HD) disease patients, and healthy age-matched controls as a means to assess the integrity of L-type channels in these neurological disorders often associated with the aging brain.
DHP and PA receptor binding parameters were not significantly altered in any of the brain regions studied in AD and PD.
However, a significant decrease in the maximal binding capacity of [3H]PN200-110 was observed in the striatum of HD patients.
Taken together, this suggests that DHP and PA binding sites associated to L-type Ca2+ channels are mostly preserved in AD and PD brains. Accordingly, the use of DHP- and PA-related drugs in these neurological disorders should not be hindered by deficits in their related Ca2÷ channel binding proteins.
Intracellular Ca2+ signaling is fundamental to neuronal physiology and viability. Due to its ubiquitous roles, disruptions in Ca2+ homeostasis are implicated in diverse disease processes and have become a major focus of study in multifactorial neurodegenerative diseases such as Alzheimer’s disease.
Azepine and Diazepine Based Peptide Mimics for AD
In the last years, there is an increasing evidence of the implication of free radicals and reactive oxygen
species in a variety of diseases and pathophysiological events including inflammation, cancer, myocardial
infarction, arthritis and neurodegenerative disorders [47]. Azepine [48] based drug candidates a key role in
this effort. Reactive Oxygen Species (ROS) have been suggested to play a key role in the pathophysiology
of myocardial reperfusion injury. Peptide isosteres have been useful for demonstrating that presenilin [49]
is the catalytic subunit of -secretase and probing the active site. Presenilin-1 and -2 Are Molecular Targets
for g-Secretase Inhibitors [50].
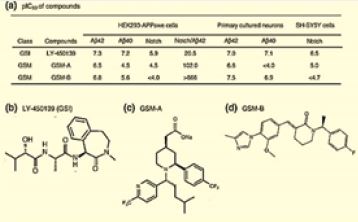
Gamma-secretase inhibitors for Alzheimer’s disease [51]: balancing efficacy and toxicity. Based on this hypothesis, compounds that inhibit gamma-secretase, one of the enzymes responsible for forming Aβ, are potential therapeutics for AD. Preclinical studies clearly establish that gamma-secretase inhibitors can reduce brain Aβ in rodent models [52]. The discovery of Begacetate [53], was giving new hope to AD patients.
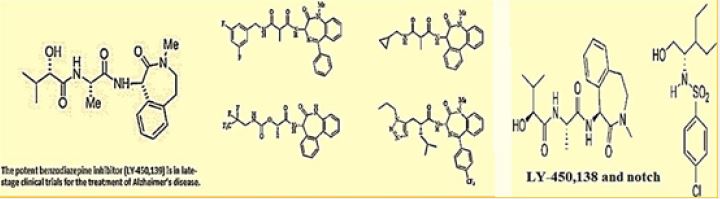
There is a potential role of antimicrobial peptides in the early onset of Alzheimer’s disease. In fact, one of the promising natural product, the semagacestat is a modified peptide compound. It is one of the most advanced candidates in respect to the therapy of Alzheimer’s. Semagacestat is a gamma-secretase inhibitor for the potential treatment of Alzheimer’s disease [54]. γ-Secretase modulators do not induce Aβ-rebound and accumulation of β-C-terminal fragment [55].
Although there was published a few US patent on Azepine derivatives as gamma-secretase inhibitors, we have prepared [56] an analog based on a pentapeptide chain (KAAAL) and a - turn mimic the benzodiazepine unit. The patent does not mention Diazepine derivatives as gamma-secretase inhibitors. We checked the antibacterial activity to both the DHP based and the Benzodiazepine based peptidomimetic which both were active antibacterial agents.
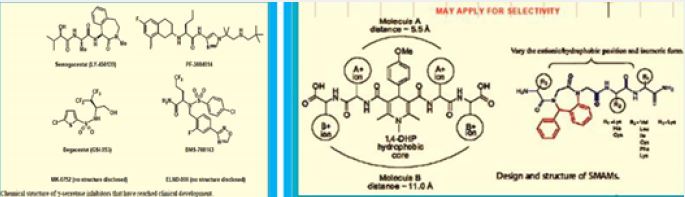
Astra-Zeneca explored the g-secretase inhibition and noch inhibition with some diazepine based peptide mimics follows:
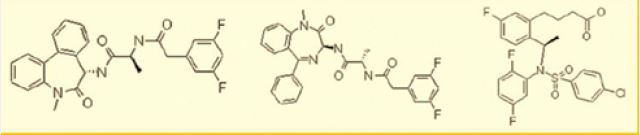
Small Molecules are Targeted as Neurodegeneration Diseases Future Drugs [4]
There are new prospects and strategies for drug target discovery in Neurodegenerative disorders. The
future of the neurological therapeutic development depends upon effective disease modification strategies
centered on carefully investigated targets. Pharmaceutical research endeavors that probe for a much deeper
understanding of disease pathogenesis, and explain how adaptive or compensatory mechanisms might be
engaged to delay disease onset or progression, will produce the needed breakthroughs. Below, we discuss the
prospects for new targets emerging out of the study of brain disease genes and their associated pathogenic
pathways. We describe a general experimental paradigm that we are employing across several mouse models
of neurodegenerative disease to elucidate molecular determinants of selective neuronal vulnerability. We
outline key elements of our target discovery program and provide examples of how we integrate genomic
technologies, neuroanatomical methods, and mouse genetics in the search for neurodegenerative disease
targets.
The general approach tend to encumber many neurological brain diseases and seek the diagnosis and cure of the very essential dysfunction sites with decoy therapeutics.

Focus is on the general situation that combines many diseases and efforts are placed in the understanding of the biological processes leading to the grim situation an effort to eliminate the factors that take the undesired biologically imposed process and modify it in such a manner that successive events which lead to the appearance of the diseases will be eliminated or slowed down.
With AD, Diabetes, CJD, and more, Aβ aggregation is the first target. The stacking of the Aβ molecules to fibrils and further is a crucial stage in the development of the diseases.
Conclusions
The efforts towards the effective curing efforts of neurodegenerative diseases (also called sometimes
conformational diseases), and in the front-line Alzheimer’s and Parkinson’s diseases is gaining a new
perspective. The joining of Diabetes and neuro-degradation as leading concept for the better understanding
and more accurate and profound understanding is moving the focus of attention to new ideas based on
peptides and their surrogate to block pathogenic processes in the development of biological agents that
destroy the brain cells, Aside insulin, Amylin [59] has become an additional digestive peptide that is
considered as target for exploration and exploiting the biological processes for the design of novel, peptide
mimics, anti neuro-degenerative agents.
The new hope in the efforts toward new treatment, quantitative early diagnostics, mechanisms of action and anti-infective treatments or a new serum for vaccination, are the aspiration of many which will finally improve and might solve the long-standing problems in neuro-degradation.
Bibliography


Hi!
We're here to answer your questions!
Send us a message via Whatsapp, and we'll reply the moment we're available!