Biography
Interests
Dr. Shimon Shatzmiller*, Dr. Inbal Lapidot, Dr. Galina Zats, Dr. Ludmila Buzhansky & Dr. Rami Krieger
Department of Chemical Sciences, Ariel University, Israel
*Correspondence to: Dr. Shimon Shatzmiller, Department of Chemical Sciences, Ariel University, Israel.
Copyright © 2018 Dr. Shimon Shatzmiller, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
WHO Report [1]
• YAntimicrobial resistance (AMR) threatens the effective prevention and treatment of an ever-increasing range of infections caused by bacteria, parasites, viruses and fungi.
• AMR is an increasingly serious threat to global public health that requires action across all government sectors and society.
• Without effective antibiotics, the success of major surgery and cancer chemotherapy would be compromised.
• The cost of health care for patients with resistant infections is higher than care for patients with nonresistant infections due to longer duration of illness, additional tests and use of more expensive drugs.
• In 2016, 490 000 people developed multi-drug resistant TB globally, and drug resistance is starting to complicate the fight against HIV and malaria, as well.
Antimicrobial resistance happens when microorganisms (such as bacteria, fungi, viruses, and parasites) change when they are exposed to antimicrobial drugs (such as antibiotics, antifungals, antivirals, antimalarials, and anthelmintics). Microorganisms that develop antimicrobial resistance are sometimes referred to as “superbugs.”
As a result, the medicines become ineffective, and infections persist in the body, increasing the risk of spread to others.
Opening Words
The human health problem that annoys the population of earth these days due to the resistance and
character-change of many bacteria strands due to horizontal gene transfer is threatening public health,
The nosocomial infections situation is alarming, one of 25 entering a healthcare facility gets infected and
one of nine dies from the infection. Antibiotic resistance is a growing public health problem. The United
Nations recently acknowledged this as “one of the biggest threats to modern medicine [2],” dedicating a highlevel meeting to the issue at the 2016 General Assembly [3]. The infectors are now everywhere in the public domain, but mainly in lavatories, restaurants, recreation parks for example, everywhere, and this overflow of infectors is the next even more alarming stage of the pandemic. There is an urgent need to harness forces for the combat with the microbes, with the hope to win this battle. Hand wash may help, but more thorough
hygiene is needed, even in the soil. The current regulatory environment is a further factor constricting
research and development of antibacterial agents by large and small pharmaceutical companies. Regulatory
authorities are less prepared to accept adverse side-effects with antibiotics than other classes of therapeutic
agents and demonstration of superiority is required for new antimicrobials. Both these factors are likely to
contribute to a further decline in pharmaceutical industry involvement in the discovery and development of
new antibacterial and antiseptic agents.
Recently, Macrophages express a variety of receptors for pathogen associated molecular pathways (PAMPs) such as lipopolysaccharide (LPS) and mannose receptors. These enable the recognition and phagocytosis of a large repertoire of pathogens, and the subsequent generation of inflammation to eradicate the infection [4].
Challenge
One major world problem, the microbe’s pandemic, should be addressed biblically. Erase the old and look for new, state of the art antimicrobial [5] and antiseptic agent.
We, humans, have adapted to live in harmony with different microorganisms throughout evolution - the microbiome [6]; this balanced symbiotic relationship can sometimes shift and allow pathogenic bacteria to blossom and cause infections. In the struggle for survival, a complex mechanism involving many key components assists in the elimination of these infectious agents.
The problem’s focuses are many [7], here we mention three target areas: Health (Hospital Acquired Infections, HAI), Food sterilization and Agriculture [8] (Livestock, soil sterilization).
Introduction
Healthcare-associated infections (HAIs)-infections patients can get while receiving medical treatment in a
healthcare facility-are a major, yet often preventable, threat to patient safety. Together with health care and
public health partners, CDC is working to bring increased attention to HAIs and prevention.
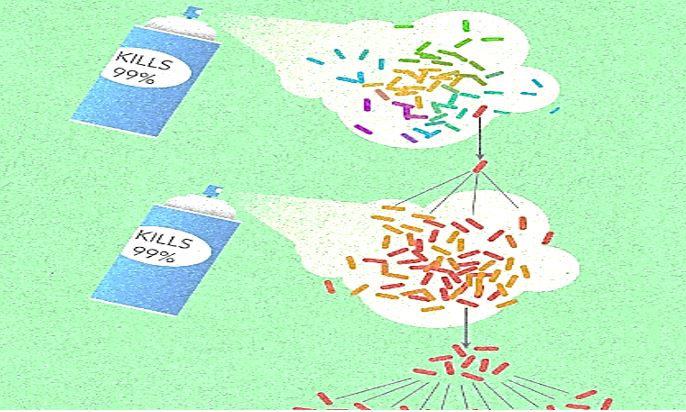
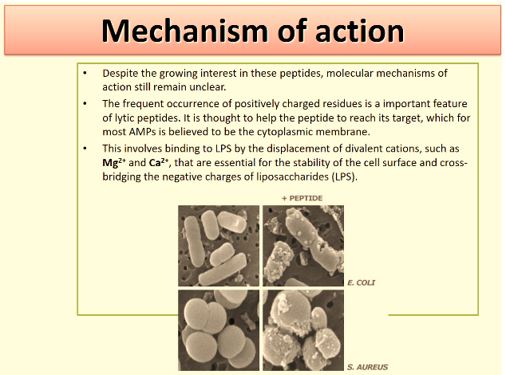
The advantage of antimicrobial peptides is the generality of their mechanism of action, which involves either compromising the bacterial membrane integrity or disrupting essential components inside the cells. This differs from the specific receptors targeted by conventional antibiotics which allow the pathogenic bacteria to develop resistance more rapidly. Furthermore, antimicrobial peptides are fast-acting and biodegradable, which alleviates the current concern over residual antibiotics in the environment. In addition to their direct microbicidal activities, these host defense peptides are particularly attractive because of the multiple activities that are associated with many members of this family. These include the regulation of the innate and adaptive immune systems, inflammation and wound healing, and additional anti-infective activities such as being antifungal, antiviral, antiparasitic or anticancerous.
However, This perception of the bioactivity of Amp and presumably their surrogates mat be too over estimated. The case with Gramicidin may contradict it.
Antimicrobial agents have changed human and animal health systematism by revolutionizing our weapons in the war against infectious disease, resulting in improved survivability [9]. There was a time where the perfect antimicrobial drug “The magic bullet a search for the perfect drug” was considered a n achievable target [10]. For both human and animals. However, this health triumph has been tempered by the subsequent realization that bacterial populations can quickly modify them to resist [11].
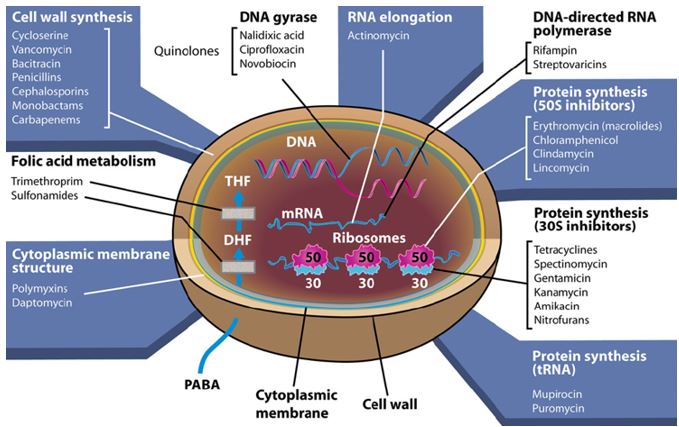
Frequent antibiotic use over long periods of time puts selective pressure on bacteria and causes resistance to spread. When an antibiotic is used to treat a typical bacterial infection, most bacteria are killed. Sometimes, however, a bacterium with an advantage life. This bacterium can then reproduce and pass its advantage on, creating many more antibiotic-resistant bacteria.
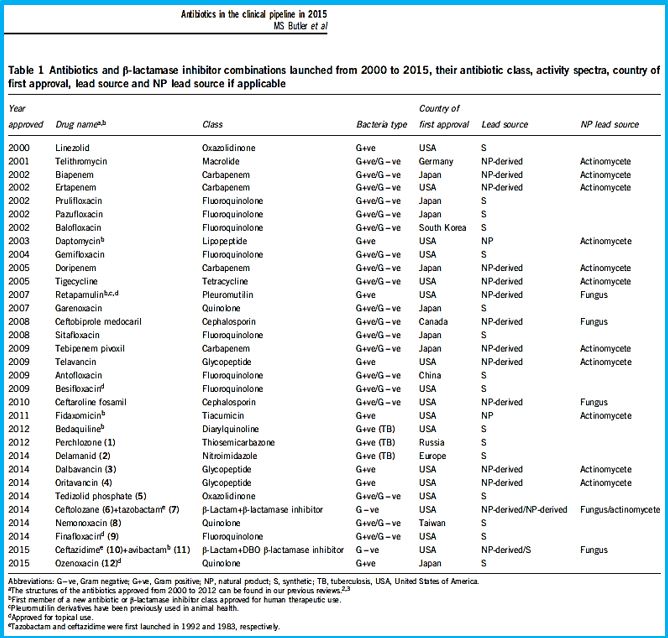
The short list in the table above represents some of the newest antibiotic drugs approved for antiinfection treatment. Please notice the poor yields in antibacterial agents in the last decades, only a score of novel drugs was found and approved [12], do not provide weapons to fight infections in healthcare installations (nosocomial infections) and give only little hope to the millions suffering from the fact6 that they do not provide remedy to those that are infected with the resistant microbes and in most cases, must die while treated in hospitals.
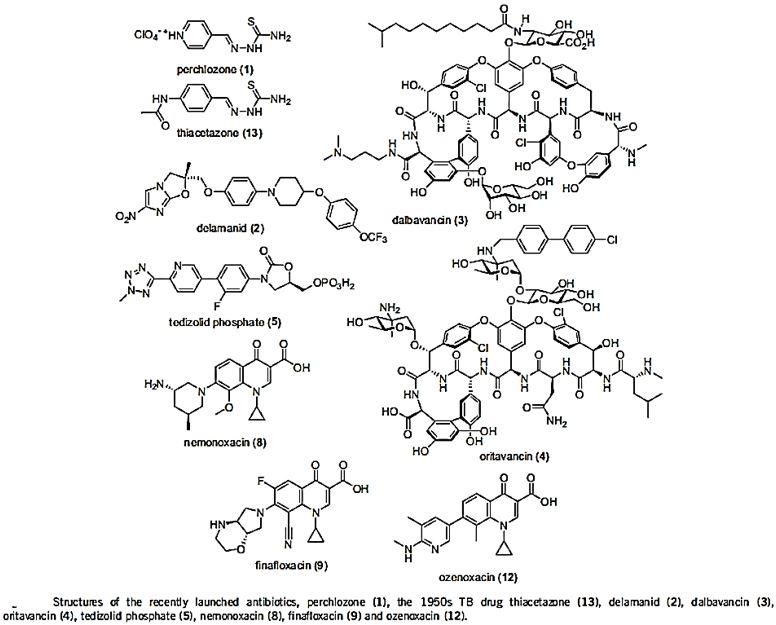
One of the reasons is that even the most modern findings in the area are impotent in the killing of bacteria when resistant strands are the matter.
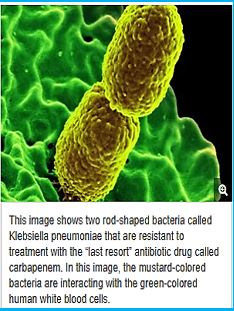
CRE are a type of bacteria that are highly resistant to many antibiotics. They have earned the name “killer bacteria” and “nightmare bacteria” for good reason. The death rate for those infected with CRE, or carbapenem-resistant Enterobacteriaceae, (The Enterobacteriaceae are a large family of Gram-negative bacteria) is greater than 40 percent, according to the California Department of Public Health (CDPH), and could be as high as 50 percent, according to the Centers for Disease Control (CDC). Infections usually occur in hospitals, nursing homes and other health care settings, the CDC said. Members of the general population are generally not at risk.
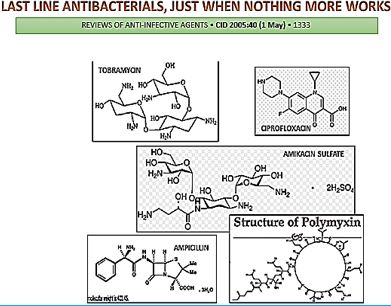
The emergence of drug-resistant strains of major pathogenic bacteria such as Staphylococcus aureus is an increasingly serious public health concern [13]. To evade bacterial drug resistance mechanisms, new effective chemotherapeutic agents, which have novel mechanisms of action as well as different cellular targets compared with conventional antibiotics, need to be developed [2]. The shortage of new antibiotics to combat multidrug-resistant (MDR) strains has led to a renewed interest in polymyxins [14]. Polymyxins are active against MDR Gram-negative bacteria such as Pseudomonas aeruginosa, Klebsiella pneumonia, Acinetobacter baumannii, and Enterobacter species [15]. Early reports described high incidences of nephrotoxicity and neurotoxicity during polymyxin therapy [9], and the use of polymyxins was replaced in the 1970s by antibiotics considered to be less toxic. However, recent studies showed that polymyxins have acceptable effectiveness and are considerably less toxic than originally reported. Cationic am Colistin (polymyxin B) was re-introduced as “last resort” for treating patients with acute infection based on drugresistant bacteria, However established that the cyclic peptide is very harmful to central organs like heart, liver, kidney brain and is a lethal substance in about 60% of its use causes mortality Polymyxins, a group of polypeptide antibiotics that consists of 5 chemically different in 1947. Only polymyxin B and polymyxin E (Colistin) have been used extensively worldwide in Colistin was discovered in 1949 and was non-ribosomally synthesized by Ba5]. Coli of colistimethate sodium in 1959. However, them due to multidrug-resistant (MDR), gram-negative bacteria in patients with cystic fibrosis. However, the emergence of bacteria resistant to most classes of commercially available antibiotics and the shortage of new antimicrobial agents with activity against gram-negative microorganisms have led to the reconsideration of polymyxins as a valuable therapeutic option.
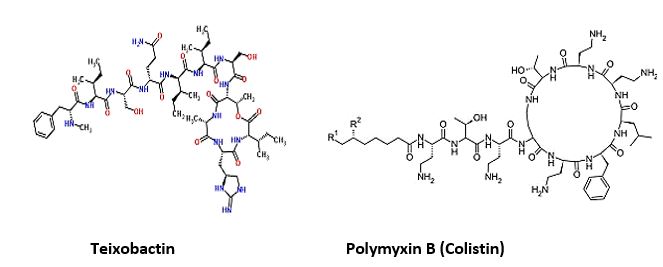
The novel antimicrobial peptide-based Teixobactin identified [17,18]. Recently, Scientists have discovered an
antibiotic capable of fighting infections that kill hundreds of thousands of people each year, a breakthrough
that could lead to the field’s first major new drug in more than a quarter-century. The new agent that was
isolated, identified and synthesized, gives great hope to medicine regarding the combat withy resistant
bacteria. Teixobactin and some of its analogs were synthesized by a number of research groups. This awakes
the hopes that are centered around the following:
1. Selectivity preferred eradication, Gram+ or Gram-.
2. Effective eradication is of all bacteria regular and resistant.
3. Low toxicity to humans.
Not only one target is attacked by Teixobactin, but multiple targets, and they are all lethal. For bacteria, it will be very hard to modify this target, especially this part of the molecule that’s bound by Teixobactin.
Until the discovery of Teixobactin, the following were the top antibiotics in the clinic:
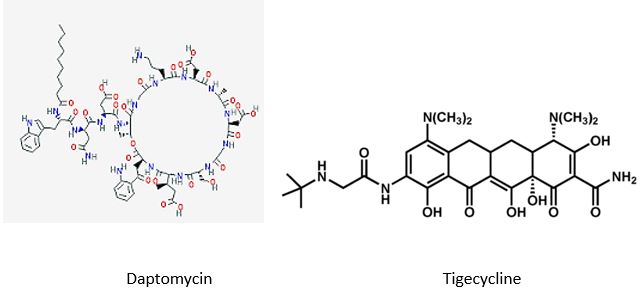
The last major new antibiotic, daptomycin, was discovered in the 1980s by Eli Lilly & Co. After being abandoned in early testing, Antibiotic-resistant bacteria kill at least 700,000 people a year, per a U.K. government review. Unchecked, those infections could lead to 10 million more deaths a year by 2050, the report found.
Tigecycline [19] is the one of newest antibiotic to have been released. It has been on the market for years. It This includes activity against gram-positives, gram-negatives except for P. Aeruginosa, many anaerobes and many atypical pathogens as well as several resistant pathogens. Because it is not vulnerable to many of the resistance mechanisms that affect the beta-lactam antibiotics, it has potential utility in those types of situations. It has been approved for use in complicated skin and skin structure infections, as well as for treatment of complicated intra-abdominal infections due to susceptible pathogens. These are the only 2 indications that are approved right now. It has very interesting pharmacokinetics and a long half-life. It will be dosed, after a load, twice a day, and it has a rather low serum concentration, but very good tissue distribution. Tigecycline is available as an intravenous solution. The primary side effects or adverse reactions that have been seen in clinical trials have been gastrointestinal symptoms, usually nausea.
The hunt for new antibiotics is benefiting from recent technological advances which make it easier to rapidly comb the DNA of different soil organisms, which are otherwise hard to raise in a petri dish [20].
The soil is a good place to look for new organisms because it is here that bacteria naturally compete for resources and use a range of exotic chemical compounds to kill each other.
The paladin chemical, identified by Dr Sean Brady and his laboratory at Rockefeller University, New York, works by attacking a fundamental step in bacterial growth, essentially interfering with a major building block that the bacteria use to build and repair their outer membrane.
In their study, published as a letter in the journal Nature Microbiology, the authors cautioned that it is only effective against one group of bacteria - the gram positives, which include MRSA. The majority of these skin infections are caused by Staphylococcus aureus, which can include the drug-resistant MRSA bacteria, the researchers explained in background notes [21]. Most staph skin infections are easily treated with antibiotics or by draining the infection. Some staph bacteria such as MRSA (methicillin-resistant Staphylococcus aureus) are resistant to certain antibiotics, making infections harder to treat.
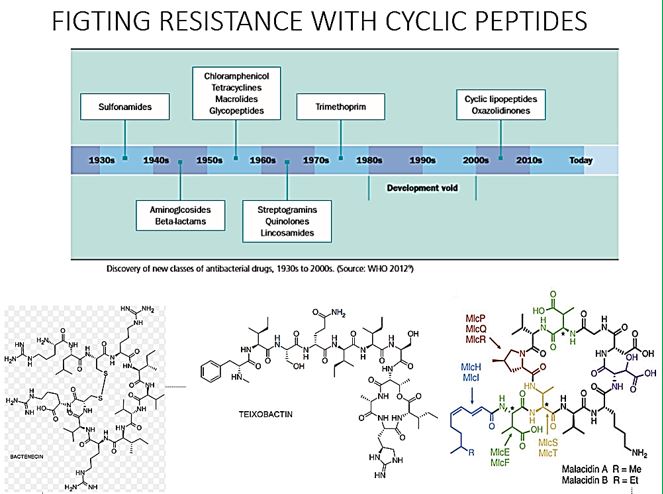
Cyclic peptides are leading the current anti-resistant antimicrobials. Bactenecin, Teixobactin [22], and the Milacainides are the most active that eradicate resistant bacteria strands. The finding new antibiotics to treat gram-positive infections like MRSA was good news but could not address the most pressing need. Our concern is the so-called gram-negative bacteria which are difficult to treat and where resistance is on the increase.
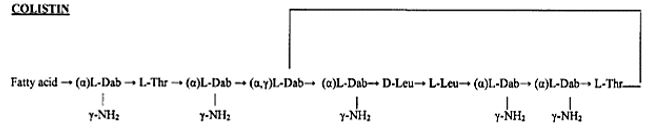
Colistin [23]: The Revival of Polymyxins for the Management of Multidrug-Resistant Gram-Negative Bacterial Infections [24].
Gram-negative bacteria cause pneumonia, blood sepsis [25] and urinary tract infections as skin infections. Twenty-four patients (mean age 44.3 years, mean Acute Physiology and Chronic Health Evaluation II score 20.6) received 26 courses of colistin. Clinical response was observed for 73% of the treatments. Survival at 30 days was 57.7%. Deterioration in renal [26] function was observed in 14.3% of 21 patients who were not already receiving renal [27] replacement therapy, but in only one case did this deterioration have serious clinical consequences. Colistin was discovered in 1949 and was nonribosomally synthesized by Bacillus polymyxa subspecies colistinus Koyama [28]. Colistin was initially used therapeutically in Japan and in Europe during the 1950s and in the United States in the form of colistimethate sodium in 1959 [6]. However, the intravenous formulations of colistin and polymyxin B were gradually abandoned in most parts of the world in the early 1980s because of the reported high incidence of nephrotoxicity. Subsequently, the intravenous use of colistin was mainly restricted during the past 2 decades for the treatment of lung infections due to multidrug-resistant (MDR), gram-negative bacteria in patients with cystic fibrosis. However, the emergence of bacteria resistant to most classes of commercially available antibiotics and the shortage of new antimicrobial agents with activity against gram-negative microorganisms have led to the reconsideration of polymyxins as a valuable therapeutic option. Currently applied Fosfomycin or Colistin may cause renal damages, it is therefore demands less damaging antibiotic agents. It seems that we need new antibiotics to treat this class.
Carbapenem-Resistant Enterobacteriaceae (CRE) Infection
CRE infections [29] are most commonly seen in people with exposure to healthcare settings like hospitals and
long-term care facilities, such as skilled nursing facilities, and long-term acute care hospitals. In healthcare
settings, CRE infections occur among sick patients who are receiving treatment for other conditions. Patients
whose care requires devices like ventilators (breathing machines), urinary (bladder) catheters, or intravenous
(vein) catheters, and patients who are taking long courses of certain antibiotics are among those at risk for
CRE infections.
In recent years, carbapenem resistance among Enterobacteriaceae has dramatically increased and represents an important threat to global health.
Some CRE bacteria have become resistant to almost all available antibiotics and can be deadly-one report cites they can contribute to death in up to 50% of patients who become infected. The Therapeutic options for carbapenem-resistant Enterobacteriaceae infections are limited.
Some reports have investigated the clinical efficacy of intravenous Fosfomycin [30] in patients with CRE infections. Michalopoulos et al. reported 11 cases of ICU patients having CRKP infections who were treated with intravenous fosfomycin in combination with other antibiotics; the clinical and microbiological outcome was good with an all-cause hospital mortality of 18.2%, and no adverse events were reported. More recently, Pontikis et al. investigated clinical outcome of 48 fosfomycin-treated (mainly in combination with colistin or tigecycline) ICU patients having XDR fosfomycin-susceptible P. Aeruginosa (n D 17) and K. Pneumoniae (n D 41) carbapenemase-producing isolates; the authors reported a survival rate of 54.2%, evidence of bacterial eradication in 56.3% of cases, and development of fosfomycin-resistance in 3 cases. The N-marcolide antibiotic Azithomyxcin is applied in cases of an allergic reaction or a type of diarrhea caused by Clostridium difficile is possible.
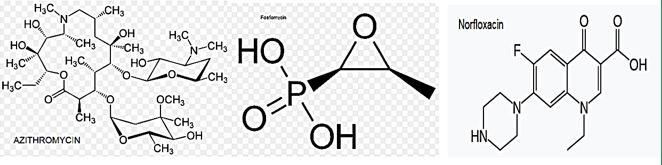
Fosfomycin (also known as phosphomycin or phosphonomycin and the trade names Monurol and Monuril) is a broad-spectrum antibiotic produced by certain Streptomyces species, although chemical synthesis can make it.
As a single dose, fosfomycin is more convenient than multiple-dose therapy Norfloxacin, for the same antibacterial efficacy.
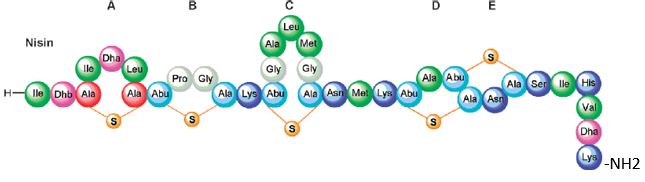
Bioactive Macrocyclic Peptides and Peptide Mimics [31a-c], AIB -based peptide backbone as scaffolds for helical peptide mimics, Antibiotics like Nisin and other peptides and mimics were tested as potent agents for resistant bacteria.
Nisin is a bacteriocin produced by Lactococcus lactis subsp. Latics that exhibits a broad inhibitory spectrum against gram-positive bacteria, including bacterial endospores [32a,b]. Recent studies have shown that the spectrum of nisin activity can be extended to include gram-negative bacteria. Application of nisin in combination with the chelating agent EDTA results in inhibition of Salmonella species and other gramnegative bacteria.
One of the approaches is to learn about the potential application of short peptide mimics like β-turn mimics, on the recognition with perspective to apply this if future drug design. The appearance of β-turns in protein interaction is by far more common than that of other, like γ-turns. Non-covalent [33,34] interactions between the β-turn-mimics and some receptors on the cell wall of bacteria may supply enough energy differences that may allow differentiation between various bacterial transmembrane cell wall receptors [35] due to the receptor and β-turn mimic interactions. The interactions of some β-turn mimics with many classes of proteins which vary in their secondary structure (β-sheets, globular) have been found to rely on the interaction between β-turn mimics and the proteins.
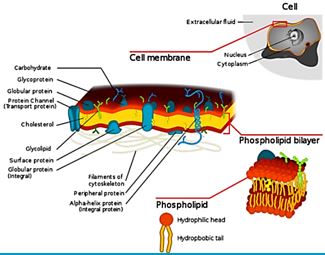
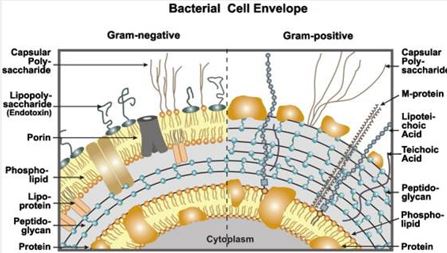
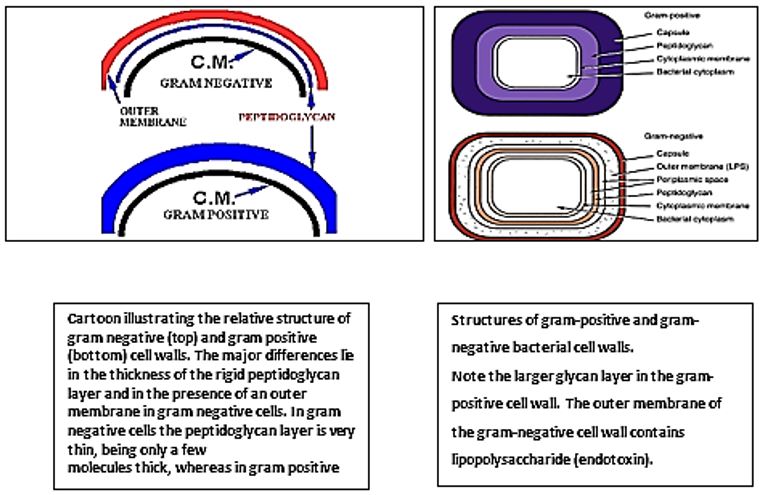
Bacteria Cell Wall and Proteins [36]
Many variants of β-turn mimic, one count nine β-turn types [37], have been applied so far in this area of research. It has been reported that Surrogate at the β-turn domain of Gramicidin S increase the biological activity [38]. One can read about benzodiazepines, β-turn mimic Hot═Tap for example [39].
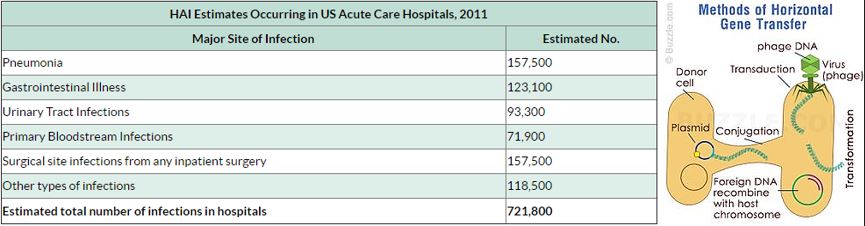
The lack of production and introduction of the newer and effective antibiotic/antibacterial drugs in clinical practice in the post-antibiotic golden age [40] has seen an increase in the emergence of the resistant pathogenic bacterial infections creating a significant problem in the global health of humankind. The situation today is that in 2011, an estimated 722,000 [41] patients contracted an infection during a stay in an acute care hospital in the US; 205 Americans die from hospital-acquired infections (hospital-acquired infections HAI, nosocomial infections [42]) every day.
Compounding the problem of antimicrobial-drug resistance is the immediate threat of a reduction in the discovery and development of new antibiotics [43,44].
CIt is now known that many different Gram-positive and Gram-negative pathogens communicate via the production and sensing of small, diffusible signal molecules, to coordinate virulence determinant production [45]. Horizontal genes transfer enables the transfer of resistance from one sort of bacteria to another either by Transformation, involves uptake of short fragments of naked DNA by naturally transformable bacteria. Transduction, involves transfer of DNA from one bacterium into another via bacteriophages. Conjugation which involves transfer of DNA via sexual pilus and requires cell-to-cell contact [46] (see cartoon below). Hunting the nightmare bacteria [47], Therefore, this event, now termed quorum sensing, represents a novel therapeutic target offering the opportunity to attenuate virulence, and thus control infection, by blocking cell-to-cell communication.
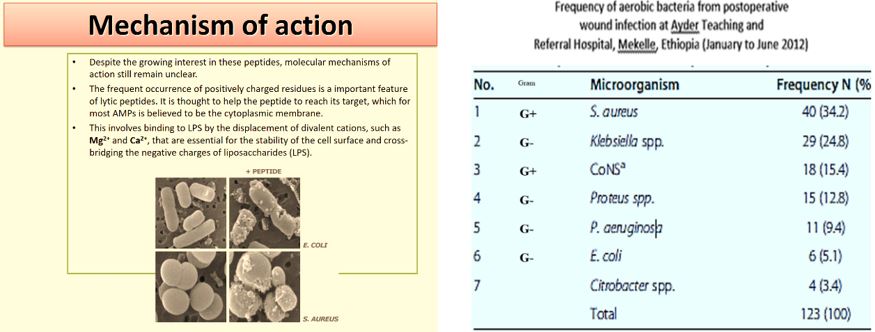
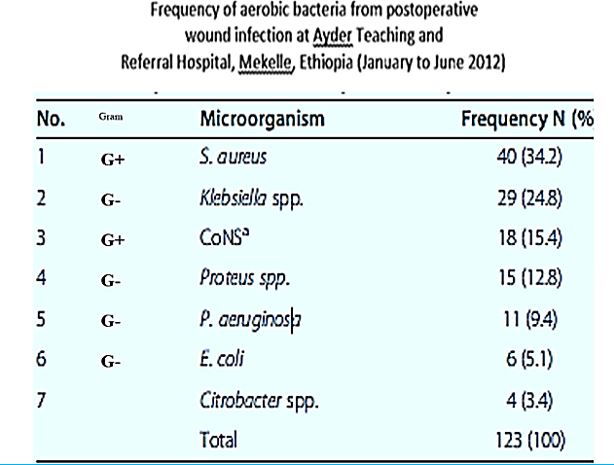
The frequency of aerobic bacteria from postoperative wound infection at Ayder Teaching and Referral Hospital, Mekelle, Ethiopia (January to June 2012) [48].
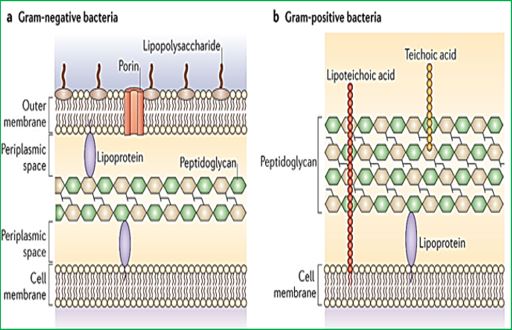
Wall teichoic acids are found only in certain Gram-positive bacteria (such as staphylococci, streptococci, lactobacilli, and Bacillus spp); so far, they have not been found in gram-negative organisms. Teichoic acids are polyol phosphate polymers, with either ribitol or glycerol linked by phosphodiester bonds; their structures are illustrated in Figure 2-9. Substituent groups on the polyol chains can include D-alanine (ester linked), N-acetylglucosamine, N-acetylglucosamine, and glucose; the substituent is characteristic for the teichoic acid from a bacterial species and can act as a specific antigenic determinant. Teichoic acids are covalently linked to the peptidoglycan. These highly negatively charged polymers of the bacterial wall can serve as a cation-sequestering [49].
Assessing Compound Permeability in Gram-Negative Bacteria to Enable Rational Drug
Design
In Gram-negative bacteria, the envelope is a sophisticated barrier protecting the cell against external toxic compounds. Membrane transporters, e.g., porins or efflux pumps, are main filters regulating the internal accumulation of various hydrophilic molecules. Regarding bacterial susceptibility towards antibacterial
agents, membrane permeability is part of the early bacterial defense. The bacterium manages the translocation
process, influx and efflux, to control the intracellular concentration of various molecules. Antibiotics and
biocides are substrates of these mechanisms and the continuing emergence of multidrug resistant isolates is a
growing worldwide health concern. Different strategies could be proposed to bypass the bacterial membrane
barrier, comprising influx and efflux mechanisms, in order to restore the activity of antibiotics [50].
Accessing drug targets in the cytoplasm of Gram-negative pathogens is difficult because of the permeability barrier that limits the accumulation of promising antibiotics to effective levels within the cell. The ability to measure compound permeation and accumulation in Gram-negative bacteria is potentially an enabling component of structure-activity-relationships which guide rational drug design and optimization. Strategies addressing these challenges and the development of enabling technologies will be discussed.
While antibiotic resistance is increasing rapidly, drug discovery has proven to be extremely difficult. Antibiotic resistance transforms some bacterial infections into deadly medical conditions. A significant challenge in antibiotic discovery is designing potent molecules that enter Gram-negative bacteria and also avoid active efflux mechanisms. Critical analysis in rational drug design has been hindered by the lack of effective analytical tools to analyze the bacterial membrane permeability of small molecules. We design, fabricate, and characterize the nanofluidic device that actively loads more than 200 single bacterial cells in a nanochannel array. We demonstrate a gigaohm seal between the nanochannel walls and the loaded bacteria, restricting small molecule transport to only occur through the bacterial cell envelope. Quantitation of clindamycin translocation through wild-type and efflux-deficient (ΔtolC) Escherichia coli strains via nanofluidic-interfaced liquid chromatography-mass spectrometry shows higher levels of translocation for wild-type E. coli than for an efflux-deficient strain. We believe that the assessment of compound permeability in Gram-negative bacteria via the nanofluidic analysis platform will be an impactful tool for compound permeation and efflux studies in bacteria to assist rational antibiotic design.
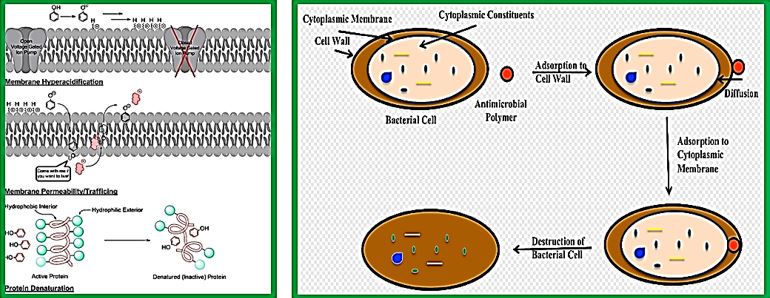
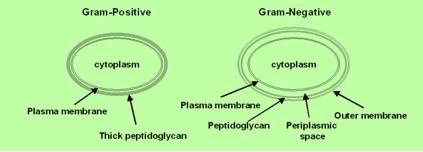
The eradication of a Gram-positive cell is demanding the crossing of one cell envelope, the outer membrane Blue in diagram) whereas the Gram-negative cell eradication demands the damaging of both the outer and inner membranes.

The transport of solutes through to the inner or cytoplasmic membrane of bacteria usually takes place through specific active transport systems that require energy [51]. The transport of the agents from the on the outer membrane organized carpet to its destination in the inner cell wall is different regarding the two sorts of bacteria. We presume that the penetration of the molecule into the membrane of the bacteria is different in the two types: Whereas penetration [52] of the peptide mimic to the Gram-negative bacteria needs a great energetic [53] effort due to the crowded situation used by stacking it with membrane proteins and lipo-proteins [54], demanding a high energy track for both types of the surrogates where the slight change in energy demand is too small compared to the overall penetration energy [55].
Recently [56], evidence was provided that killing takes place only when bacterial cell membranes are completely saturated with AMPs. This condition is achieved for all bacteria. However, Since the in Gramnegative bacteria the outer membrane are crowded, packed with various proteins (up to 50% of the total membrane weight), compared of only 15% in the S-layer (surface layer) [57] in gram-positive, it demands more energy for saturation in gram-negative than in gram-positive.
The introduction of the N-CH3 unit to the Penta peptides surrogates stiffens the structure thereby causes an increase in energetic demand for saturation. The fraction of this energy in Gram-negative is smaller than in Gram-positive due to membrane packing composition. It is easier for the N-CH3 to penetrate the outer membrane in gram-positive bacteria and in Gram-negative. Since saturation is achieved in gram-positive with fewer energy demands, the Gram-positive compared to gram-negative are eradicated in preference (through “snorkeling” [58] for example).
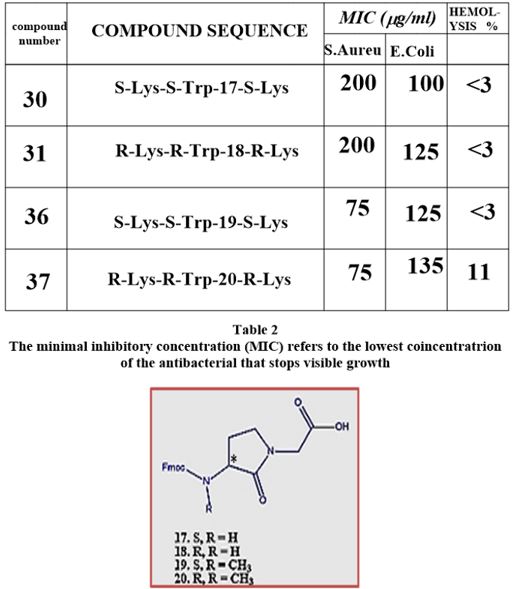
Conclusion from the experiment of N-methylation
The Gram-positive bacteria are easier to penetrate [59] since the outer membrane is poor in membrane
proteins. In such circumstance, small energy changes can become significant for the travel of the agent into
the outer membrane. The N-CH3 are less flexible and therefore penetrates easier to the membrane. Finally,
after the surrogates settle in the inner part of the outer membrane, the Lys ε-amine unit can snorkel [60]
out and disintegrate the membranes of both Gram-positive and Gram-negative bacteria. The interactions
of an AMP with the membrane cannot be explained by a sequential amino-acid pattern or motif; rather,
they originate from a combination of physicochemical and structural features [61] including size, residue
composition, overall charge, secondary structure, hydrophobicity and amphiphilic character [62].
The interactions of an AMPs surrogates with the membrane cannot be explained only by a particular sequential amino-acid pattern or motif; rather, they originate from a combination of physicochemical and structural features [63] including size, residue composition, overall charge, secondary structure, hydrophobicity and amphiphilic character [64,65]. Also, interactions with the many components that furnish the architecture of the membranes are crucial. From our experiment, we conclude that the venerability of bacteria may depend on small structural variation in the composition of the biocide [8].
New Drugs Needed
Human medicine has reaped the bene fits of antibiotics for the past 80 years, but without urgent action, the
utility of these agents will be drastically minimized. To limit the spread of resistance, physicians must use
antibiotics judiciously and apply infection-control procedures consistently. These measures alone, however,
will not be sufficient. New antibiotics are desperately needed. To address this emerging crisis, the medical
community, governments, pharmaceutical companies, and public health agencies must all work together.
Only a coordinated and committed response can slow the rising tide of multidrug-resistant bacteria.
Abstract
The emergence of multidrug-resistant Gram-negative bacteria that because nosocomial infections is a growing problem worldwide. Colistin was first introduced in 1952 and was used until the early 1980’s for the treatment of infections caused by Gram-negative bacilli. In vitro, colistin has demonstrated excellent activity against various Gram-negative rod-shaped bacteria, including multidrug-resistant Pseudomonas aeruginosa, Acinetobacter baumannii, and Klebsiella pneumoniae. Recent clinical findings regarding colistin activity, pharmacokinetic properties, clinical uses, emerging resistance, toxicities and combination therapy have been reviewed. Recent approaches to the use of colistin in combination with other antibiotics hold promise for increased antibacterial efficacy. It is probable that colistin will be the ‘last-line’ therapeutic drug against multidrug-resistant Gram-negative pathogens in the 21st century.
Introduction
The polymyxin lipodecapeptides colistin and polymyxin B have become last resort therapies for infections
caused by highly drug-resistant Gram-negative bacteria. Unfortunately, their utility is compromised by
significant nephrotoxicity and polymyxin-resistant bacterial strains.
We owe much of that, of course, to antibiotics. The discovery of Prontosil, the first synthetic modern antibiotic, earned Gerhard Domagk the Nobel Prize in 1939. Massproduced penicillin earned Alexander Fleming, Ernst Boris Chain, and Howard Walter Florey one in 1945. It is hard to overstate how much less of a threat of infectious disease pose to us today. But we take antibiotics for granted. We use them inappropriately and indiscriminately. This has led many to worry that our days of receiving benefits from them are numbered. In a recent report published in Lancet Infectious Diseases, scientists discovered colistin-resistant E coli in 21percent of slaughtered pigs in China. They found isolates in 15 percent of meat sold from those animals in retail sites. They even detected resistant E. Coli in more than 1percent of hospitalized patients.
While we can quibble about the exact cost of bringing a new drug to market, we can all agree that it’s a lot of money. Drugs in the United States are profitable when they are sold in great volume or when they are very expensive. Antibiotics, as a class of drug, provide a poor return on investment for pharmaceutical companies. They face low-priced and generic competition. Any breakthrough drug will almost certainly be held in reserve for only the most resistant cases, meaning there’s not a huge immediate market for it when a company still has exclusivity.
It’s this new drug investment that might bring the most hope. For many years, spending on antibiotic resistance research was flat. Only in the last year or two have both the president and Congress seemed especially interested in pushing for more funding. New antibiotics are a public good, not necessarily an area for private rewards. Huge public support for researching new classes of antibiotics will be necessary to combat this growing threat.
As a sage observer once noted, though, bringing new antibiotics to market without changing how we use them is kin to providing alcoholics with a finer grade of brandy. For this reason, most new funding goes toward prevention, infection control and managing antibiotic use. The best outcome is preventing infections through vaccination or public health measures so that we improve human health without increasing resistance to antibiotics.
Recent years research afforded some peptide-based agents that are the most effective agents in the combat against resistant microbes. Research based on such compounds may give rise to a new trend in the quest of remedy to the vicious killers that transform modern medicine to better treatment of Hospital Acquired Infection (HAI), or the nosocomial pandemic. Bacterial sepsis kills millions of people every year. In the United States, patients hospitalized for septicemia or sepsis are more than eight times as likely to die during their hospitalization compared to those hospitalized for other diagnoses, with over 200,000 deaths in 2008. Over one-third of patients with sepsis treated in an intensive care unit die in the hospital. Sepsis is the most expensive condition treated in U.S. hospitals, costing more than $20 billion in 2011. Sepsis is most commonly caused by Gram-positive (G+ve) Staphylococcus aureus and Streptococcus pyogenes and Gramnegative (G-ve) Klebsiella spp., Escherichia coli, and Pseudomonas aeruginosa [5]. Unfortunately, there has been an ominous rise in highly drug-resistant G-ve bacteria that can overcome almost every known antibiotic, threatening a return to a “antibiotic” era if new therapies are not discovered. The current clinical pipeline is mainly populated by G+ve drug candidates [6]. One possible approach to antibiotic development is to revisit antibiotics discovered in the “golden age” of antibiotic discovery (the 1950s−1970s) to see if they can be optimized or improved.
The upper box depicts the structures of the resurgence of “VETERAN” but highly bioactive (Gramicidin [66], 1940, Polymyxins 1947) that can eradicate the most aggressive resistant bacteria strands, However, these are highly toxic to humans.
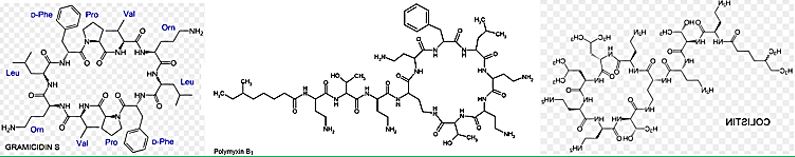
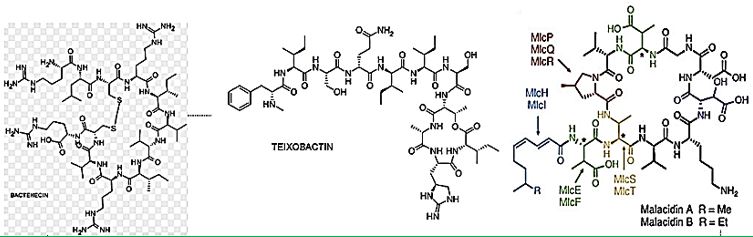
Cyclic [67] peptides are leading the current anti-resistant antimicrobials. Bactenecin, Teixobactin, and the Milacainides are the most active that eradicate resistant bacteria strands.
The finding new antibiotics to treat gram-positive infections like MRSA was good news but could not address the most pressing need. Our concern is the so-called gram-negative bacteria which are difficult to treat and where resistance is on the increase.
The quest for selective agents for the eradication of resistant bacterial strands, preferable of Gram negative bacteria, led to the discovery that cyclic peptides may penetrate and disrupt bacterial membrane by selfassembly to stacks of rings:
The rapid emergence of bacterial infections that are resistant to many drugs underscores the need for new therapeutic agents. Here were port that six-and eight-residue
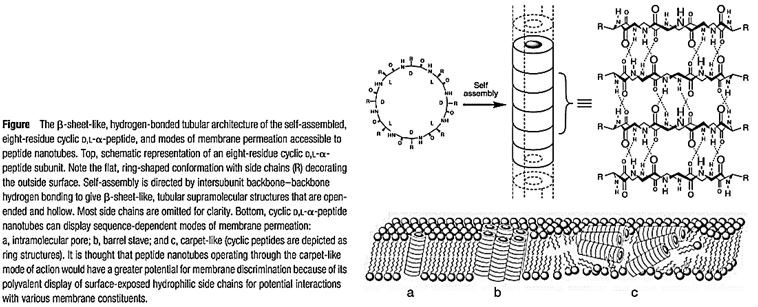
Cyclic D,L-α-peptides act preferentially on Gram-positive and Gram-negative bacterial membranes compared to mammalian cells, increase membrane permeability, collapse transmembrane ion potentials, and cause rapid cell death. The by the high efficacy observed against lethal methicillin effectiveness of this class of materials as selective antibacterial agents are highlighted -resistant Staphylococcus aureus infections in mice. Cyclic D, L-a-peptides are proteolytically stable, easy to synthesize, and can be derived from a potentially vast membrane-active sequence space. The unique abiotic structure of the cyclic peptides and their quick bactericidal action may also contribute to limit temporal acquirement of drug-resistant bacteria. The low molecular weight D, L-a-peptides offer an attractive complement to the current arsenal of naturally derived antibiotics and hold considerable potential in combating a variety of existing and emerging infectious diseases [68].
Cyclic polypeptides are about 1/3 of the types of antimicrobial agents. Namely Ansamycins, Streptogramins and the Lipopeptides. Colistin and other Polymyxins are considered as Lipopeptides.

Polymyxins have become the drug of choice for treatment of multidrug-resistant gram-negative bacilli infections, simply because these pathogens are only susceptible to either aminoglycosides and polymyxins, or polymyxins only. Furthermore, there is no new antibiotic in the pipeline that targets these difficult-to-treat infections.
Materials and this article serve to give a summary of polymyxins from the currently available literature, highlighting relevant clinical studies and information that help to guide informed prescription of polymyxins, should the need arise. However, there are substantial information gaps that needed to be filled urgently, to preserve the clinical utility of this very last line of antibiotic. As we can see in the structures, the commercial drug Colistin is a mimic of Polymyxin B. where a D-Phe was replaced by D-Leu. The prodrug is, in fact, the salt Colistimethate- (Na+). Modifications [69] of-of both Colistin and Polymyxins are designed and synthesized, aiming at the elimination of the neurological and nephrological side effects, with only some success.
The situation of the patient once they are attacked with a resistant strand of bacteria is extremely hazardous, even when Colistin or Fosfomycin are used as therapeutic agents. Therefore, the in vitro Activity of Polymyxin B in Combination with Various Antibiotics against Extensively Drug-Resistant Enterobacter cloacae with Decreased Susceptibility to Polymyxin B may provide some hope for safer recovery [71]. Against extensively drug-resistant (XDR) Enterobacter cloacae., combination antibiotic therapy may be the only option. We investigated the activity of various antibiotics in combination with polymyxin B using timekill studies (TKS). TKS were conducted with four nonclonal XDR E. cloacae isolates with 5 log10 CFU/ ml bacteria against maximum, clinically achievable concentrations of polymyxin B alone and in two-drug combinations with ten different antibiotics. A hollow-fiber infection model (HFIM) simulating clinically relevant polymyxin B and tigecycline dosing regimens were conducted for two isolates over 240 h. The emergence of resistance was quantified using antibiotic-containing (3× MIC) media. Biofitness and stability of resistant phenotypes were determined. All XDR E. Cloacae isolates were resistant to all antibiotics except for polymyxin B (polymyxin B MIC, 1 to 4 mg/liter). All isolates harbored Metallo-β-lactamases (two with NDM-1, two with IMP-1). In Conclusion. In the study, researchers found that polymyxin B in combination with tigecycline is a promising treatment against XDR E. Cloacae
However, it must be highlighted that prolonged and indiscriminate use, especially if employed at suboptimal doses, can result in the emergence of resistance strains. Future studies investigating polymyxin B combinations in animal models and in patients, with additional studies to characterize the virulence of the resistance strains, will be required.
Polymyxin B has resurged in recent years as a last resort therapy for Gram-negative multidrug-resistant (MDR) and extremely drug resistant (XDR) infections. Understanding newer evidence on polymyxin B is necessary to guide clinical decision making [72]. Polymyxin B has resurged in recent years as a last resort therapy for Gram-negative multidrug-resistant (MDR) and extremely drug resistant (XDR) infections. Understanding newer evidence on polymyxin B is necessary to guide clinical decision making. Here, we present a literature review of polymyxin B in Gram-negative infections with an update on its pharmacology.
Polymyxins are one of the frontline antibiotics which have been revived in the past few years [73]. The resurgence of polymyxins in GNB infections especially the MDR Pseudomonas Aeruginosa, Acinetobacter Baumannii, and Klebsiella Pneumoniae has been the significant Growing use of polymyxins in GNB infections was identified and perceived in consensus for optimization on the clinical use of polymyxins “The Prato Polymyxin Consensus”. Given increasing use of polymyxin B in clinical settings, in this review, we provide current literature evidence of polymyxin B use as monotherapy and combination therapy for MDR and XDR Gram-negative infections.
Chemistry of Colistin
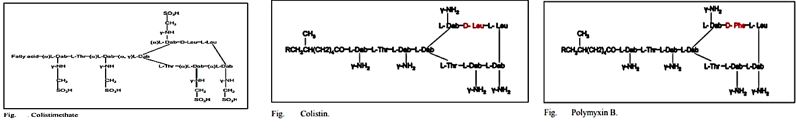
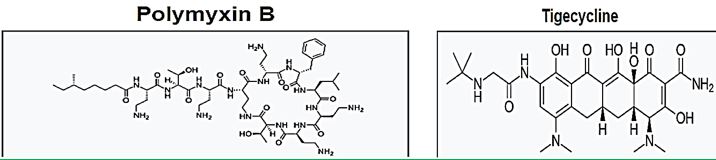
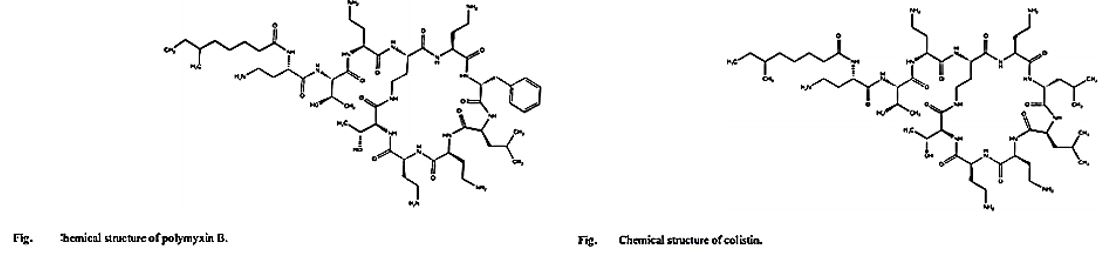
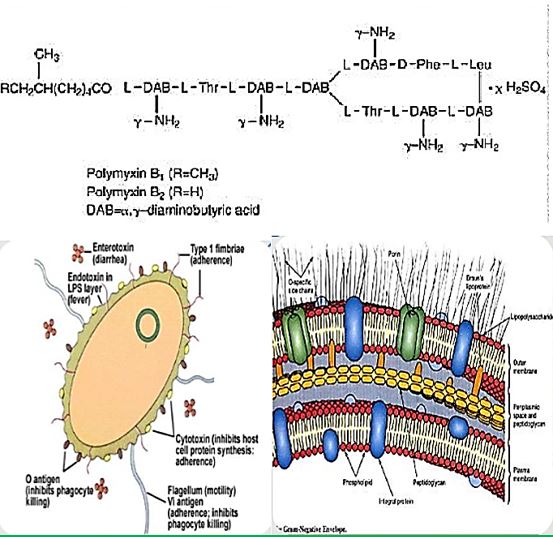
The emergence of bacterial resistance toward currently employed antibiotics has led to the reuse of ‘abundant’ antibiotics such as polymyxin B (PMB) toward multidrug resistance strains of P. Aeruginosa, Enterobacter, Serratia and E. Coli [74]. PMB is a naturally occurring cationic cyclic lipopeptide, isolated from Bacillus polymyxa [75], highly bactericidal to Gram-negative bacteria and considered one of the most efficient cellpermeabilizing compound [6]. PMB is composed of a positively charged cyclic peptide ring and a fatty acid containing tail (Fig. 1). The cyclic part is a seven-member amino acid ring containing four 2,4-diaminobutyric acids (Dab) residues, one Thr residue and a hydrophobic segment of DPhe-Leu. The C-terminal carboxylic function of Thr9 forms an amide bond with the -amino group of Dab3 (Fig. 1).

The linear N-terminal region is composed of two Dab and one Thr residues together with a 9 or 8 carbon fatty acid, i.e. 6-methyl octanoic [PMB1] and 6-methyl heptanoic acid [PMB2], respectively, forming a long hydrophobic tail. It was shown that both the positive charges and the acylated linear tail are crucial for antibacterial activity [76]. The unique structural features of the cationic peptide are required for binding specifically to lipid A of Gram-negative bacterial lipopolysaccharide (LPS) allowing the acylated hydrophobic tail to penetrate through the bacterial outer membrane [77]. PMB is unique among antibiotics in that it has potent anti-endotoxin activity in addition to its antibacterial activity. This property could be beneficial to patients who are experiencing Gram-negative bacterial sepsis and endotoxin-mediated shock. PMB inhibits LPS-induced macrophage production [78] of interferon-, tumor necrosis factor (TNF) and interleukin-1 and 6 (IL-1 and IL-6). In vivo, PMB can act against most Gram-negative septicemia and prevents endotoxin lethality in several animal models [79]. The therapeutic applications of PMB are very limited, however, because of its relatively high toxicity and as a consequence it is used mainly for topical treatment.
Colistin is a multicomponent polypeptide antibiotic that is mainly composed of colistin A, and colistin B. PMB and colistin (polymyxin E) are secondary metabolite nonribosomal peptides that share a similar primary sequence with the only difference being at position6, which is replaced by D-Phe in PMB and D-Leu in colistin (Figure 2).
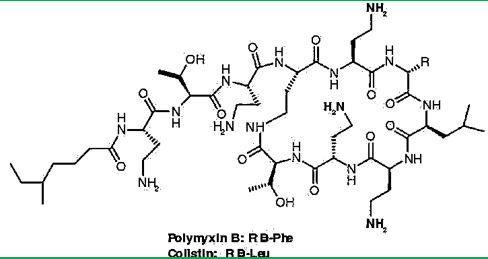
Structure of polymyxin B and colistin. Polymyxin B and colistin (polymyxin E) share a similar primary sequence with the only difference being at position 6, in which D-Phe in polymyxin B is replaced by and D-Leu in colistin.
Colistin sulfate and CMS are the two forms of colistin. CMS is produced by the reaction of colistin with formaldehyde and sodium bisulfite, which leads to the addition of a sulfomethyl group to the primary amines of colistin [80]. Although CMS is the form administered parenterally, it undergoes conversion invivo to form colistin, which is responsible for the antibacterial activity, and thus CMS should be considered as an inactive prodrug.
The importance of the N-terminal fatty acyl segment for the antimicrobial properties of polymyxins first became evident when polymyxin nonapeptides were identified [81]. Although PMB and colistin nonapeptides lack any direct antimicrobial activity, they possess the same ability to bind lipopolysaccharide (LPS) with an important specificity and perturb the outer membrane (OM) integrity to sensitize Gram-negative bacteria to hydrophobic antibiotics that are not normally active.
Previous studies of the Nα fatty acyl structure-activity relationships (SARs) of PMB and colistin component peptides isolated from cultured strains did not provide any clear and meaningful SAR data [82]. The most comprehensive NαSAR data have been reported by Sakura and colleagues [83], wherein purified PMB or colistin were converted to nonapeptides by treatment with S-ethyl trifluorothioacetate and used as the starting material.
Recently, Sakura and colleagues have reported a new series of Nα analogs derived by acylation of the tetrakis (Nγ-Troc)-PMB or -colistin nonapeptides with various hydrophobic acids and aliphatic or hydrophobic ring structures. The Nα analogs were tested for their LPS binding affinity and antimicrobial activity against Escherichia Coli, and Salmonella Enterica and P. Aeruginosa. Thus, cyclohexylbutanoyl, 4-biphenylacetyl, and 1-adamantaneacetyl-Nα analogs led to comparable activities concerning the parent compounds (PMB and colistin), with an improved LPS binding affinity [84].
Tsubery et al. adopted a synthetic approach by using a combination of solid-phase linear chain elongation methodology and subsequent cyclization after release [85]. Owing to the presence of a long hydrophobic chain, [Ala]6-PMB was expected to increase the potent antimicrobial activity; nevertheless, neither of the oligoclonal Nα analogs had significantly better activity than the control product PMB. The N-terminus of the other pair was substituted with the hydrophobic Fmoc group. This result demonstrates that the hydrophobicity of the Nα substituent group greatly influences the outcome of antimicrobial activity and the inherent acute toxicity. Finally, N-terminal modifications of PMB nonapeptide have been shown to possess high antibacterial activity and significantly reduced toxicity.
Polymyxin B, is a clinically important member of the polymyxin group of antibiotics that are cyclic peptides highly potent against gram-negative bacteria Nephrotoxicity and neuromuscular blockade are serious side effects associated with this class of antibiotics, and hence their clinical usage is primarily as topical agents. 2 Polymyxin B contains five free T-amino groups from the ~,7-diaminobutyric acid (DAB) units and these functionalities are attractive handles that can be used for chemical modifications/derivatizations aimed at improving the therapeutic ratio of the parent compound. Several such modifications of Polymyxin B reported previously, have shown that monoacylation of Polymyxin B retained the antibacterial potency whereas diand polyadenylation caused an appreciable loss in potency. However, the chemistry used in all of those studies was uncontrolled and did not establish which of the amino groups of Polymyxin B were derivatized.
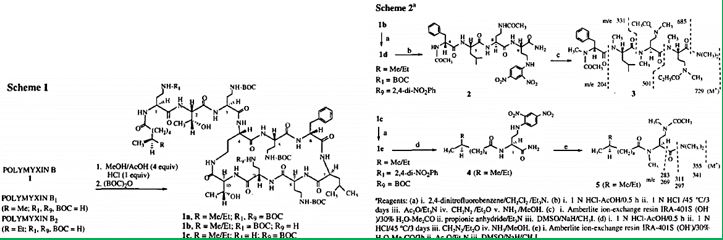
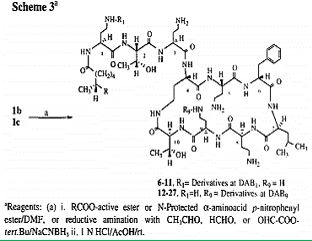
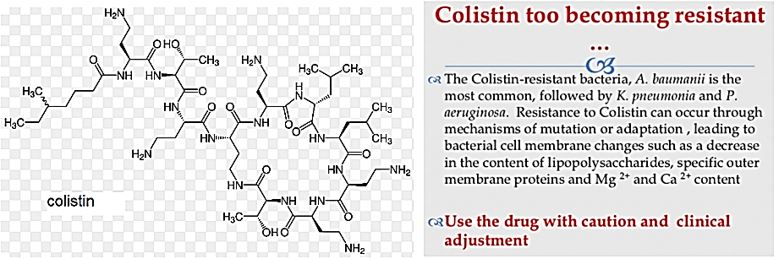
Here us the methodology [86] to selectively functionalize the DAB ~ or the DAB 9 amino group of Polymyxin B. The structures of the precursor tetra-BOC-polymyxin B derivatives lb and lc was established using a chemical degradation sequence which utilizes a new DNP deprotection method. In contrast with previous studies ~ on the chemical modifications of Polymyxin B, our study identifies the site of the functionalization of the parent antibiotic. Of the twenty-two polymyxin B derivatives reported here, compound 24 was found to have an improved toxicity-activity profile in vivo. The chemical modifications of colistin may become one of the major tools to develop the new generation of modified polymyxins, those that will eradicate the Colistin resistant bacteria [87]. Namely, Colistin-resistant K. Pneumonia, A. Baumannii, and P. Aeruginosa pathogens may be encountered in clinical practice, in association with inappropriate colistin use. To prevent this phenomenon, colistin should be used judiciously, given that treatment options for colistin-resistant Gram-negative bacteria are limited. Antibiotic resistance is a growing crisis and a grave threat to human health. It is projected that antibiotic-resistant infections will lead to 10 million annual deaths worldwide by the year 2050.
Among the most significant threats are carbapenem-resistant Enterobacteriaceae (CRE), including carbapenem-resistant Klebsiella Pneumonia (CRKP), which lead to mortality rates as high as 40 to 50%. Few treatment options are available to treat CRKP, and the polymyxin antibiotic colistin is often the “last-line” therapy. However, resistance to colistin is increasing. This is concerning because Klebsiella is a more common cause of infection than Enterobacter, and these isolates were carbapenem-resistant, which means that they might actually be treated with colistin. To our knowledge, this type of heteroresistant Klebsiella has not been observed in the United States before. The bacterial isolates came from urine samples from two patients in Atlanta-area hospitals as part of the nationwide Multi-site Gram-Negative Surveillance Initiative, part of the CDC-funded Emerging Infections Program. Heteroresistance is caused by a minor subpopulation of resistant bacteria that are genetically identical to the rest of the susceptible bacteria, and it means that bacterial resistance to particular antibiotics is harder to monitor.
The “last resort drugs”, the modified Polymyxines, suffer from many drawbacks, including deadly side effects and recently, as mentioned, the resistance of various microbial strands. In particular, the cyclic cationic peptides polymyxin B and colistin, which are specific for Gram-negative bacteria, have been used as ‘last resort’ antimicrobials. Before the 1980s, these drugs were known for their renal and neural toxicities; however, new clinical practices and possibly improved manufacturing have made them safer to use. Previously suggested to primarily attack the membranes of Gram-negative bacteria and to not easily select for resistant mutants, recent research exploring resistance and mechanisms of action has provided new perspectives.”
According to the news editors, the research concluded: This review focuses primarily on the proposed alternative mechanisms of action, known resistance mechanisms, and how these supports the alternative mechanisms of action [88].
Antibiotic resistance among pathogenic bacteria is an ever-increasing issue worldwide. Unfortunately, very little has been achieved in the pharmaceutical industry to combat this problem. This has led researchers and the medical field to revisit past drugs that were deemed too toxic for clinical use. The cyclic cationic peptides polymyxin B and colistin [89], which are specific for Gram-negative bacteria, have been used as “last resort” antimicrobials. Before the 1980s, these drugs were known for their renal and neural toxicities; however, new clinical practices and possibly improved manufacturing have made them safer to use. Previously suggested to primarily attack the membranes of Gram-negative bacteria and to not easily select for resistant mutants, recent research exploring resistance and mechanisms of action has provided new perspectives. This review focuses primarily on the proposed alternative mechanisms of action, known resistance mechanisms, and how these support the alternative mechanisms of action.
Using Colistin
The rapid emergence of bacterial infections that are resistant to many drugs underscores the need for new therapeutic agents.
Cyclic peptides are leading the current anti-resistant antimicrobials. Bactenecin, Teixobactin, and the Milacainides [90] are the most active that eradicate resistant bacteria strands.

The finding new antibiotics to treat gram-positive infections like MRSA was good news but could not address the most pressing need. Our concern is the so-called gram-negative bacteria which are difficult to treat and where resistance is on the increase.
The History of Colistin in Short
Colistin was one of the first antibiotics to be developed in the early 1950s [91], but it wasn’t used to treat serious infections in human medicine because it is very toxic. All antibiotics have side effects which affect different individuals to different extents, but few of them are as severe as those associated with colistin, which can cause kidney and neurological damage that is occasionally irreversible.
It’s hardly surprising, therefore, that drug companies originally assumed there would be no problem if colistin were developed for use in livestock production, as products like Coliscour and Colibird. One of its main uses has simply been to prevent or treat diarrhea in piglets caused by E. coli bacteria which spread very quickly on intensive farms and especially in piglets weaned at 3-4 weeks of age because their immune systems do not function fully until they are about eight weeks of age.
But it is also used to treat and prevent E. Coli and salmonella infections in veal calves and poultry. E. Coli blood poisoning infections affect about 40,000 people a year in the UK every year and kill about 7,000 of them, including from time to time newborn babies which become infected in the birth canal [92].
In March 2016 [93], the CDC reported that “superbugs” - bacteria that are highly resistant to antibiotics - are responsible for one out of seven infections caught in general hospitals, causing an annual estimate of 80,000 in the U.S. Several types of bacterial infections have evolved to ‘deadly invader’ status, and it’s partially our fault. Among the 18 superbugs identified by the CDC are salmonella, drug-resistant tuberculosis, and a staph bacterium known as MRSA (Methicillin-resistant Staphylococcus aureus). These bugs are gaining ‘super’ status because we’ve been careless and stupid with antibiotics like Colistin, rendering them useless, if not dangerous. In other words, we’re helping create deadly types of bacteria. The revival of Colistin is the rehabilitation of a renal [94] damaging agent; it is the best drug to apply when there is no other choice.
Colistin is also one of only four antibiotics which can be added to the drinking water of egg-laying hens suffering where there is no requirement to observe any withdrawal period at all. Eggs on sale can come from hens right in the middle of antibiotic treatment, and chicken [95] can be slaughtered for human consumption just 24 hours after being treated with colistin. One of the most disconcerting things about the regulation of farm antibiotics, is that the legal withdrawal periods - the number of days from zero up to 40 (or more in some cases) after treatment when meat, milk or eggs are not supposed to enter the food chain, is based only on the level of antibiotic residues and takes no account the levels of antibiotic resistance.
Colistin is a complex polypeptide antibiotic composed mainly of colistin A and B. It was abandoned from clinical use in the 1970s because of significant renal and, to a lesser extent, neurological toxicity. Colistin is increasingly put forward as salvage or even first-line treatment for severe multidrug-resistant, Gramnegative bacterial infections, particularly in the intensive care setting.
Polymyxins are polypeptide antibiotics, with a primary effect of membrane-damaging due to their selective binding to the lipopolysaccharide of Gram-negative bacteria [96]. Their nephew- and neurotoxic side effects [97] limited their use, however, in the last decade the emergence of multidrug-resistant Gramnegative bacteria led to the reintroduction of polymyxins into clinical practice. This review provides an overview of the history and the latest developments of polymyxins. We describe the antimicrobial effects, pharmacodynamics, pharmacokinetics and different routes of administration. We highlight natural classic polymyxins, namely polymyxin B and E, the non-classic agent’s polymyxin M, S and T. Novel polymyxin chemical structure derivatives will be listed, that can have important therapeutically role in the future.
The emergence of multiple-drug-resistant (MDR) gram-negative microorganisms, including Pseudomonas Aeruginosa and Acinetobacter Baumannii, is a major concern worldwide [98] MDR A. Baumannii and P. Aeruginosa are important causes of nosocomial infection, and outbreaks of these microorganisms, which are resistant to most available antimicrobial agents, have been reported in burn units, intensive care units (ICUs), cancer centers, and patients with cystic fibrosis [99].
The WHO recognized the importance of colistin in anti-microbial therapy. Expert Committee on Biological Standardization (1964) decided that there was a need for an international reference preparation of colistin methane sulfonate and requested the National Institute for Medical Research, London, to obtain a suitable sample and make a preliminary evaluation of it [100].
The general mechanism [101] for the antibiotic activities of the polymyxins and octapeptins has been elucidated by research using a broad range of experimental techniques. Since 1947, when polymyxin was first isolated, there have been tremendous advances in our knowledge of membrane structure. The application of biophysical technology such as NMR, ESR, fluorescence spectroscopy, differential scanning calorimetry, and electron microscopy has been particularly valuable for studying model and biological membrane structures. It is these techniques which will provide a detailed molecular mechanism for the effects of these peptide antibiotics on membrane structure. Also, the large number of antibiotic derivatives available should be exploited more extensively for structure-function correlations. The ultimate goal is to correlate the biological properties of these peptides with their effects on the physical properties of membranes and to rationalize these events regarding lipid-peptide interactions.
The chemical synthesis was published, and a patent application was filed.
Increasing number of infection cases caused by multiresistant Gram-negative bacteria or a multidrug-resistant organism (MDRO) has become a major problem worldwide since there has been a lot of resistance to many classes of antibiotics. Mutant isolates such as fluoroquinolone-resistant and -lactamase-resistant bacteria have been commonly found, particularly in intensive care unit (ICU). During the last two decades, there has been no study of developing antibiotics in search of discovering a new type of antibiotics; meanwhile, the resistance of Gram-negative bacteria or MDRO to antibiotics is increasing. Colistin [104] or polymyxin E is an old antibiotic, which has been used since 1959 for treating infection caused by Gram-negative MDRO. It was revealed that colistin has side effects of nephrotoxicity and neurotoxicity; therefore, the use of this antibiotic was stopped, and it was replaced by other antibiotics which were effective and were considered safer at that time.
There is an increasing number of infections with multi-resistant Gram-negative (MDRO) against the available antibiotics. The availability of alternative antibiotics has not been satisfying; therefore, microbiologists are searching back to the old option, which has been proven to be effective against multi-resistant Gram negative bacteria, the old antibiotic that has been long forgotten, i.e., colistin, as an alternative treatment against Gram-negative MDRO. It is expected that colistin may have an essential and reliable role as future antibiotics for treatment of multi-resistant Gram-negative infections and as an alternative of antibiotics that have been available so far.
Here are some facts on Colistin: Colistin is a real-life saver, it is one of the most active agents in the combat with the Gram-negative resistant bacteria. Its mechanism of action disrupts both the inner and outer membranes of the Gram-negative bacteria.
Polymyxins are antibiotics, structure consisting of a cyclic peptide with a long hydrophobic tail. They disrupt the structure of the bacterial cell membrane by interacting with its phospholipids. They are produced by the Gram-positive bacterium Bacillus polymyxin and are selectively toxic for Gram-negative bacteria due to their specificity for the lipopolysaccharide molecule that exists within many Gram-negative outer membranes.
Colistin - A Polymyxin E Colistin is a cationic polypeptide antibiotic from the polymyxin family that was first introduced in 1962 but abandoned in the early 1970s because of initial reports of severe toxicities. However, a recent increase in the prevalence of multidrug-resistant (MDR) Pseudomonas aeruginosa and the lack of novel agents in development calls for a need to re-examine the role of colistin therapy in patients with cystic fibrosis.
What is Colistin? - Colistin (also called polymyxin E) belongs to the polymyxin group of antibiotics. It was first isolated in Japan in 1949 from Bacillus polymyxa var. colistin and became available for clinical use in 1959. Colistin was given as an intramuscular injection for the treatment of gram-negative infections but fell out of favor after aminoglycosides became available because of its significant side effects. It was later used as topical therapy as part of selective digestive tract decontamination and is still used in an aerosolized form for patients with cystic fibrosis.
Structure of Polymyxins. Polymyxin B Sulfate is one of a group of basic polypeptide antibiotics derived from B polymyxa (B aero porous). Polymyxin B sulfate is the sulfate salt of Polymyxins B1 and B2, which are produced by the growth of Bacillus polymyxa (Prazmowski) Migula (Fam. Bacillacea)
Colistin: The target of the antimicrobial activity of colistin is the bacterial cell membrane Colistin also has potent anti-endotoxin activity. The endotoxin of G-N bacteria is the lipid A portion of LPS molecules, and colistin binds and neutralizes LPS 7
Mechanism of Action: Bactericidal. Bind to lipopolysaccharides(LPS) & phospholipids in the outer cell membrane of G (-) bacteria Neutralize LPS & prevent pathophysiologic effects of endotoxin. Resistance is uncommon Disk diffusion method cannot be used
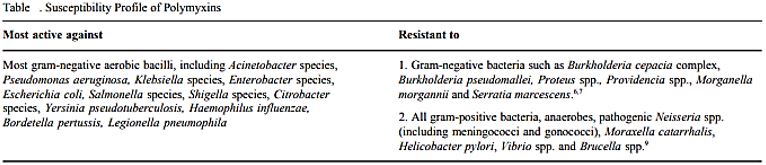
The spectrum of Activity: Pseudomonas & A. BaumanniiE. Coli, Enterobacter H. influenzaBordetella pertussis. Legionella, Klebsiella spp. Salmonella spp., Shigella spp.Stenotrophomonas maltophilia [105].
The Use of Colistin in Human and Veterinary Medicine [106]
Due to the major concerns for neuro- and nephrotoxicity [107], parenteral use of polymyxins has until recently been limited and polymyxins were mainly used for ophthalmic and topical infections. Cystic fibrosis patients have been an exception to this practice for decades, and such patients have received systemic or nebulized colistin to control lower airway bacterial infections and their complications [108]. During the last five years, two major indications have renewed the interest of polymyxins in human medicine, namely as part of surgical prophylaxis via SDD and for the treatment of MDR Gram-negative healthcare-associated infections.
For human patients, two salt forms of polymyxin E (colistin) have been widely commercially available, namely colistin sulfate and colistimethate sodium (CMS, syn colistin methanesulphonate, colistin sulphonyl methane, pentasodium colistimethanesulphate). CMS is a prodrug of colistin and is microbiologically inactive [109]. It is administered predominantly as parenteral formulations and via nebulization. After administration, CMS is hydrolyzed to colistin, which is the base component that is responsible for its antibacterial activity. Colistin sulfate is available in tablets and syrup for SDD and as topical preparations for skin infections. CMS is available for administration intravenously, intramuscularly as well as topically via aerosol (nebulization) or intraventricular administration.
Total consumption (reflecting topical, inhalational and systemic routes of administration combined) varied widely between EU/EEA countries and doubled between 2010 and 2014 following the rise in MDR Gramnegative pathogens involved in healthcare-associated infections [110].
Veterinary Medicine
Within the EU MSs, colistin and polymyxin B are authorized nationally. The main indication for colistin in veterinary medicine is an infection of the gastrointestinal tract caused by non-invasive E. Coli in pigs, poultry, cattle, sheep, goats, and rabbits. Colistin is also used in laying hens and cattle, sheep and goats producing milk for human consumption. Colistin is also active against endotoxins produced by some E. coli strains in the gastrointestinal tract. Typically, colistin products are administered orally, in feed, in drinking water, as a drench, or through milk replacer diets. Combinations of colistin with other antimicrobials are available for group treatments of food-producing animals in some EU countries. Products for parenteral and intramammary administration are also available, and infections due to Gram-negative bacteria in ruminants including endotoxemia are claimed indications. Polymyxin B is on the list of substances essential for the treatment of equidae for systemic treatment for endotoxemia (antitoxigenic effect, not antibacterial as such) associated with severe colic and other gastrointestinal diseases [111]. As in human medicine, colistin and polymyxin B have been registered for topical administration to individual veterinary patients, except for food producing animals in the case of polymyxin B, in the absence of MRLs. In companion animals, prescription eye and eardrops are available with colistin alone, or in combination with other antimicrobials. Colistin tablets are available for calves for the prevention and treatment of neonatal colibacillosis. In some EU MSs, veterinary medicinal products (VMPs) containing colistin are not on the market, i.e. not commercialized [112].
Colistin is used in aquaculture for the prevention of Gram-negative infections [113], consumption data are not available separately for this food production sector. In the Danish monitoring programme (DANMAP), details on consumption do not refer to the use of colistin in fish.
Abstract
Antibiotic-resistant bacterial infections are a major concern for public health. Phage therapy [114] Phages as bactericidal agents have been employed for 90 years as a means of treating bacterial infections in humans as well as other species, a process known as phage therapy. It has been proposed as a promising alternative to antibiotics, but an increasing number of studies suggest that both antimicrobial agents in combination are more effective in controlling pathogenic bacteria than either alone. We advocate the use of phages in combination with antibiotics and present the evolutionary basis for our claim. Also, we identify compelling challenges for the realistic application of phage-antibiotic combined therapy [115].
Introduction
The lack of production and introduction of the newer and effective antibiotic/antibacterial drugs in clinical
practice in the post-antibiotic golden age has seen an increase in the emergence of the resistant pathogenic
bacterial infections creating a significant problem in the global health of humankind. The situation today is
that in 2011, an estimated 722,000 patients contracted an infection during a stay in an acute care hospital in
the US; 205 Americans die from hospital acquired infections (hospital acquired infections HAI, nosocomial
infections [116]) every day. Compounding the problem of antimicrobial-drug resistance is the immediate
threat of a reduction in the discovery and development of new antibiotics [117].
Wall teichoic acids are found only in certain Gram-positive bacteria (such as staphylococci, streptococci, lactobacilli, and Bacillus spp); so far, they have not been found in gram- negative organisms. Teichoic acids are polyol phosphate polymers, with either ribitol or glycerol linked by phosphodiester bonds. Substituent groups on the polyol chains can include D-alanine (ester linked), N-acetylglucosamine, N-acetylgalactosamine, and glucose; the substituent is characteristic for the teichoic acid from a particular bacterial species and can act as a specific antigenic determinant. Teichoic acids are covalently linked to the peptidoglycan. These highly negatively charged polymers of the bacterial wall can serve as a cation-sequestering.
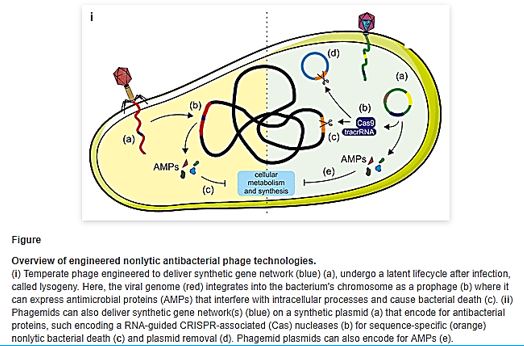
Natural phage therapies rely on causing bacterial death through rupturing cells. However, rapid bacterial lysis might result in the release of endotoxin and inflammatory mediators into the surrounding environment with adverse effects. In contrast, phages can be engineered to be bacteriostatic by deleting genes responsible for lysis (e.g., endolysin). Some tailed phages, referred to as ‘temperate,’ can undergo a latent lifecycle after infection, called lysogeny, and deliver synthetic gene networks with desirable nonlytic antimicrobial properties(). Here, the viral genome either integrates into the bacterium’s chromosome as a prophage or remains free in the cytoplasm, which is then replicated alongside the bacterium until conditions favor reactivation to produce virions. Although temperate phages are generally avoided in natural phage therapies, they have been used to deliver synthetic gene networks that can disrupt cell-cell communication between bacteria involved in biofilm formation. Or to work as adjuvants to antibiotics, such as by repressing DNA repair mechanisms or overexpressing sensitizing proteins. The drawback of these approaches is that the temperate phages would be inherently nonlethal, which one can argue would unnecessarily complicate treatment over using bacteriolytic phages. Prophage-encoded genes also carry the risk of providing a variety of benefits for their bacterial hosts. These beneficial genes are often contained within ‘moron’ elements and suggest that temperate phages have a symbiotic relationship with bacteria rather than being purely parasites as virulent (lytic) phages [118].
Patients are infected annually, and Institute for Healthcare Improvement (IHI) estimates that more than 5,000 patients die each year as a result. While most patients are treated successfully, particularly if the infection is identified early, hospital stays are often extended by an average 9.1 days, accounting for excess costs of about $20,000 per patient. The total cost burden to the US health care system from MRSA infections (Methicillin-resistant Staphylococcus aureus [119]), is a bacterial infection that is spread by direct skin-toskin contact with an infected person or by touching a contaminated surface. MRSA is estimated at more than $2.5 billion annually.
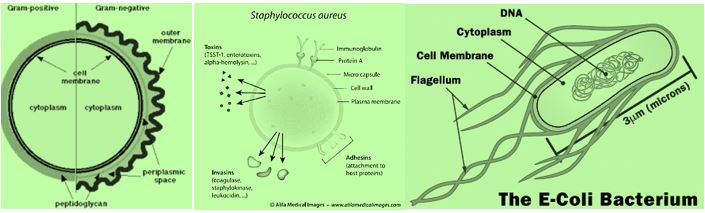
Gram-positive and Gram-negative bacteria exist everywhere (a typical post-surgery infection in the blue table below) [120,17] but pose unique threats to hospitalized patients with weak immune systems. Grampositive bacteria cause tremendous problems and are the focus of many eradication efforts, but meanwhile, Gram-negative bacteria [121] have been developing dangerous resistance and are therefore classified by the American Centers for Disease Control and Prevention (CDC) as a more serious threat. For this reason, the need for modern technologies that kill bacteria, both Gram-positive and Gram-negative, are essential to make hospitals safer for everyone. Both kinds of bacteria may cause fatal infection and should be eradicated one in the presence of the other.
The treatment of those serious bacterial infections in clinical practice is often complicated by antibiotic resistance. Therefore, there is an urgent need for innovative ideas in design and application of antimicrobial agents since bacteria gram-negative bacteria [122 ,123], develop strains that are intrinsically resistant to many antibiotics (persister) [124] strains, that are practically indifferent to all known antibiotic drugs. Furthermore, in the last decades, only two antibiotic classes with a novel mechanism of action (example is Teixobactin [125,126], a new cell wall inhibitor) have been marketed, and none of them are effective against persister Gram-negative bacteria. (The Top Ten Most Dangerous Bacteria on Earth [127]).

To date, most antibiotics are targeted at intracellular processes and must be able to penetrate the
Discussion
The misuse of antibiotics has reduced their efficacy in controlling pathogens and has led to an increase in the
number of antibiotic-resistant bacteria. As an alternative to antibiotics, bacteriophages have become a topic
of interest with the emergence of multidrug-resistant bacteria, which are a threat to public health. Recent
studies have indicated that bacteriophages can be used indirectly to detect pathogenic bacteria or directly
as biocontrol agents. Moreover, they can be used to develop new molecules for clinical applications, vaccine
production, drug design, and in the nanomedicine field via phage display.
The Use of phages in as anti-infective in therapy can be extremely successful based on the selectivity of the bacteriophages toward bacteria. Also, the mechanism of action directed at the genetic materials in the nuclei of the microbial cells is different from the cell wall disruption characteristic to most antibacterial agents, mainly antimicrobial peptides [128] and their surrogates. However, Hospital-acquired infections are; postsurgical infections are characterized by their multi-infectors blend.
We can see more than five infectors in the post-surgical infection in the Ethiopian clinic. Here we can find many sorts of infectors, Gram-negative like Klebsiella. spp. And Gram-positive S. Aureus, treatment of such wounds will apply a mixture of phages, whereas today’s treatment can apply one broad band anti-infective agents (delivered as a pill, injection, infusion) old concept of using.
The past few years have seen a significant resurgent interest in the old concept of using phage as a therapeutic tool. No doubt reports of declines both in antibiotic efficacy and in pharmaceutical company interest in developing new agents are fueling the drive for antibiotic alternatives. Although they are not without their constructive critics, whole-phage approaches may certainly become valuable anti-infective tools in certain therapeutic applications. That said, we and others envision an alternative use for phage that, although less heralded [129], focuses on their specific antibacterial components, rather than the infective virion, as the active killing agent. With the lysins emerging as the most promising antibacterial candidate, defined by their potent and specific lytic activities, we are proceeding with lysin development. Whether used topically or systemically, in humans or agriculture, as purified proteins or as forms expressed from transgenic animals or bacterial secretory systems, an increasing body of data validates the potential utility of lysins. Even beyond the lysins, phage is professional parasites of bacteria, and as such, employ an arsenal of agents to subvert host function and structure. It is only logical that a comprehensive search for new antibacterial agents would attempt to mine this vast viral pool of antibacterial functions. If it is at all possible that the ‘parts are greater than the sum’ for phage, this work is justified [130].
Indian researchers published on the test of bacteriophage versus antimicrobial agents for the treatment of murine burn treatment [131]. The widespread use of antimicrobial agents in hospital settings has led to the emergence of multidrug-resistant organisms of low virulence such as Klebsiella causing serious opportunistic infections [132]. Beside this, concerns about problems such as high cost of treatment and inability to restore initial appearance of skin have resulted in research on newer agents for the treatment of burn wounds. Bacteriophages or simply phages can be the best answer to antibiotic resistance in the treatment of bacterial infections. These phages are economical, safe, self-replicating and effective bactericidal agents. In our earlier studies, we have reported the efficacy of phage therapy in treating various infections when injected systemically in the study, the efficacy of topical application of silver nitrate and gentamicin was evaluated and compared with that of a well-characterized Klebsiella-specific phage, Kpn5, for treating K. Pneumoniae B5055 induced burn wound infection in BALB/c mice.
Phage isolation. Klebsiella-specific phage Kpn5 was isolated from a sewage sample. It’s utility in treating K. Pneumoniae B5055 induced burn wound infections on i.p. Injection has been established. In this study, phage Kpn5 was evaluated for the topical treatment of burn wound infection. Hydrogel preparation is also possible as an ointment to treat wounds. There are approaches used in the published literature on the formulation and stabilization of phage for storage and encapsulation of bacteriophage in micro- and nanostructured materials using freeze-drying (lyophilization), spray drying, in emulsions, e.g. ointments, polymeric microparticles, nanoparticles, and liposomes. As phage therapy moves forward towards Phase III clinical trials, Researchers are looking at promising new approaches for micro- and nanoencapsulation of phages and how these may address gaps in the field [133].
This study highlights the importance of silver nitrate, gentamicin and phage Kpn5 for controlling burn wound infections caused by nosocomial pathogens such as K. Pneumoniae. A single application of phage Kpn5 was found to be superior to multiple applications of silver and gentamicin in the treatment of burn wound infection caused by K. Pneumoniae B5055 in BALB/c mice. Studies using these compounds in combination to treat burn wound infection are warranted. This will not only increase the survival of the infected animals, but such a strategy will also keep a check on the development of resistant mutants, a problem frequently encountered by clinicians. In a recent study, we have demonstrated such an effect on treating biofilms of K. Pneumoniae B5055 with a combination of ciprofloxacin and phage [134].
Reports [135] indicate that appropriate administration of living phages can be used to treat lethal infectious diseases caused by gram-negative bacteria, such as Escherichia Coli, Pseudomonas Aeruginosa, Acinetobacter Baumannii, Klebsiella Pneumonia, Vibrio Vulnificus, and Salmonella spp., and gram-positive bacteria, such as Enterococcus Faecium and Staphylococcus Aureus. The phage display system and genetically modified nonreplicating phages are also effective for treatment of Helicobacter pylori and P. Aeruginosa, respectively. In addition to phage particles per se, purified phage-encoded peptidoglycan hydrolase (lysin) is also reported to be effective for the treatment of bacterial infectious diseases caused by gram-positive bacteria such as Streptococcus Pyogenes, S. Pneumoniae, Bacillus Anthracis, and group B streptococci. All phage lysins that have been studied to date exhibit immediate and strong bacteriolytic activity when applied exogenously. Furthermore, phage coded inhibitors of peptidoglycan synthesis (protein antibiotics), search methods for novel antibacterial agents using phage genome informatics, and vaccines utilizing phages or their products are being developed. Phage therapy will compensate for unavoidable complications of chemotherapy such as the appearance of multidrug resistance or substituted microbes.
However successful, Phage therapy seems to be extremely efficient in infections that are a result of one resistant type of infector. However, in cases where infections are caused by many microbes, the therapeutic efficacy of bacteriophage and the antibiotic Agent (A pharmaceutical substance) individually and in combination [136] to treat cold bacilli may be more effective.
Another clinical trial was conducted to evaluate a combination of the antibiotic enrofloxacin and intramuscularly administered bacteriophage. Both treatments individually provided effective treatments of the E. coli infection, but the synergy between the two treatments led to the total protection of the birds, thus suggesting a significant value of the combined treatment [137].
Synergy in Anti-Infective Treatment
53TMost antibiotics in human use as antibacterial agents. They are natural products, elaborated by one species of microbe (bacteria or fungi) as chemical weapons, often in times of crowding, to kill off other microbes in the neighboring microenvironment. Over the past 60-70 years most antibiotics have been discovered by screening of soil samples for such natural products that kill bacteria, including known pathogens, first on culture plates and then in animal infections. Listed by the World Health Organization as one of the top 3 threats to global public health [138], more than 2 million Americans suffer from an antibiotic resistant infection at a direct cost of over $20 billion [139] with over 23,000 dying annually [140,2].
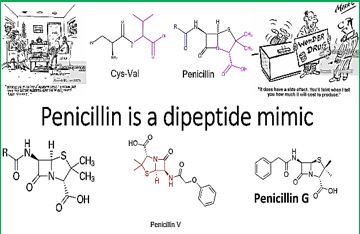
Penicillin analogs are very often used as anti-infective agents. They usually disrupt the membrane of bacterial cells and thereby curing infections. The Cell defends itself by activating some enzymes to decompose the lactam unit of the penicillins. In such circumstance, agents that inhibit the unwanted enzymatic activity are administered to protect the active penicillin agents. This is a symbiotic synergy as reflected in Augmentin.
Augmentin
amoxicillin/clavulanate potassium
Mechanism of Action: Amoxicillin inhibits bacterial cell wall synthesis, while clavulanate inhibits bacterial beta-lactamase.
Therapeutic Effect: Amoxicillin is bactericidal in susceptible microorganisms.
Clavulanate protects amoxicillin from enzymatic degradation.
Pharmacokinetics: Well absorbed from the GI tract. Protein binding: 20%. Partially metabolized in the liver. Primarily excreted in urine. Removed by hemodialysis.
Half-life: 1-1.3 hr (increased in impaired renal function).
Augmentin antibiotic appears to be a safe treatment for stroke and a good companion therapy for tPA (Tissue Plasminogen Activator - TPA).
While antibiotic resistance resulting from overuse has become a major health concern in the country, scientists noted minocycline’s use in stroke wouldn’t contribute to the problem since it will only be given for a few days. In fact, the researchers found the drug remained active in the body longer in their older stroke patients than in younger patients receiving it for other reasons, which means the patients likely will only need a single dose for three days. She and Hess have been exploring minocycline’s stroke potential for a decade [141].
The rapid proliferation of antibiotic-resistant pathogens has spurred the use of drug combinations to maintain clinical efficacy and combat the evolution of resistance. Drug pairs can interact synergistically or antagonistically, yielding inhibitory effects larger or smaller than expected from the drugs’ individual potencies. To understand how antibiotics, work and, concomitantly, why they stop being effective requires a brief look at the targets for the main classes of these antibacterial drugs. As summarized in Box 1, there are three proven targets for the main antibacterial drugs [142]:
1. Bacterial cell-wall biosynthesis;
2. Bacterial protein synthesis; and
3. Bacterial DNA replication and repair.
The molecular mechanisms [143] by which peptide antibiotics disrupt bacterial DNA synthesis, protein biosynthesis, cell wall biosynthesis, and membrane integrity are diverse, yet historically have been understood to follow a theme of one antibiotic, one inhibitory mechanism. In the past years, mechanistic and structural studies have shown a rich diversity in peptide antibiotic mechanism. Novel secondary targeting mechanisms for peptide antibiotics have recently been discovered, and the mechanisms of peptide antibiotics involved in synergistic relationships with antibiotics and proteins have been more clearly defined. In an apparent response to selective pressures, antibiotic-producing organisms have elegantly integrated multiple functions and cooperative interactions into peptide antibiotic design to improve antimicrobial success. There are Synergy and duality in peptide antibiotic mechanisms [144].
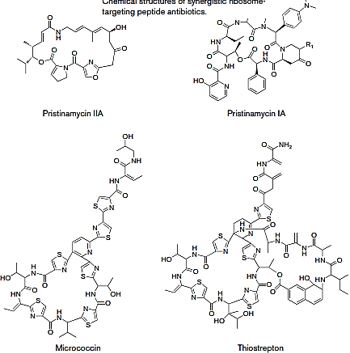
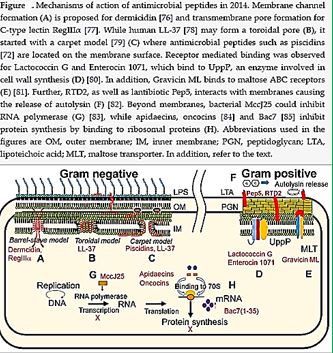
The layer of the bacterial cell wall that confers strength is the peptidoglycan, a meshwork of strands of peptide and glycan that can be covalently crosslinked (Fig. 1a). The larger the fraction of adjacent peptide strands that are connected in amide linkage by the action of a family of transpeptidases, the higher the mechanical strength to osmotic lysis. Transglycosylases act on the glycan strands to extend the sugar chains by incorporation of new peptidoglycan units from N-acetylglucosamine-b-1,4-N-acetylmuramyl-pentapeptide- pyro phosphorylundecaprenol (lipid II). Bifunctional enzymes containing both transpeptidase and transglucosylase domains are the target sites for the killing of bacteria by the b-lactam-containing penicillins and cephalosporins, which act as pseudo substrates and acylate the active sites of the transpeptidases (also termed penicillin-binding proteins or PBPs)5 (Fig. 1b). The ring-opened, penicilloylated transpeptidases deacylated very slowly, and so occupy the enzyme active sites, preventing normal crosslinking of peptide chains in the peptidoglycan layer and leaving it mechanically weak and susceptible to lysis on changes in osmotic pressure.
In addition to penicillins and cephalosporins, the vancomycin family of glycol-peptide antibiotics also target the peptidoglycan layer in the cell-wall assembly. But rather than targeting the enzymes involved in peptide crosslinking, vancomycin ties up the peptide substrate6 and thereby prevents it from reacting with either the transpeptidases or the trans-glycosylases. The net effect is the same: failure to make peptidoglycan crosslinks leads to a weaker wall that predisposes the treated bacteria to killing lysis of the cell-wall layer. The cup-shaped undersurface of the vancomycin antibiotic makes five hydrogen bonds to the D-Ala-D-Ala dipeptide terminus of each uncross linked peptidoglycan pentapeptide side chain (Fig. 1c), which accounts for the high affinity of the antibiotic for its target, both in partially crosslinked walls and in the lipid II intermediate. Because β-lactams and vancomycin work on adjacent steps substrate and enzyme, they show synergy when used in combination.
The spread of multidrug-resistant (MDR) strains of bacteria necessitate the discovery of new classes of antibacterial and compounds that inhibit these resistance mechanisms.
The transport of drugs through, and into the bacterial cell envelope, active or passive, is a major factor if the resistance of the microbes to anti-infective therapy. Reaching saturation [145] is an important factor in the agent’s efficiency as an eradicating agent.
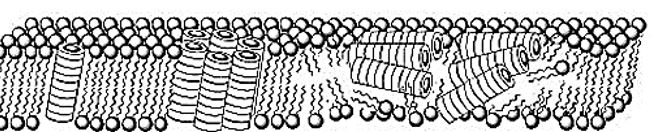
In the Drug efflux, unidirectional pumping of cytotoxic drugs, is a major mechanism of antimicrobial. multiresistance in bacteria. Although these efflux systems are usually chromosomally encoded, some are present on plasmids. Some of the efflux pumps are relatively well known: Emr and Acr system in Escherichia coli, whose outer membrane protein seems to be the multifunctional To1C; the mix efflux system described in Pseudomonas aeruginosa and ABC-type in Gram-negative bacteria. Also, the role of efflux in Grampositive bacteria are reviewed including Bacillus, Staphylococcus and Streptomyces.
Functional Synergy between Antimicrobial Peptoids and Peptides against Gram-Negative Bacteria. Antimicrobial peptides (AMPs) are integral components of innate immunity and are typically found in combinations in which they can synergize for broader-spectrum or more potent activity. Previously, scientists reported peptoid mimics of AMPs with potent and selective antimicrobial activity. Using checkerboard assays, they demonstrate that peptoids and AMPs can interact synergistically, with fractional inhibitory concentration indices as low as 0.16. These results strongly suggest that antimicrobial peptoids and peptides are functionally and mechanistically analogous [146].
The most widely used methods to treat bacterial infection are antibiotic therapies. However, traditional antibiotics are becoming less efficient because of the development of drug-resistant bacterial strains [147]. It is of great urgency to develop new and potent antibacterial materials. Reported antibacterial agents include antimicrobial peptides [148], bacteriophages [149], silver [150], carbon-based materials [151] and cationic polymers [152]. Positively charged cationic antimicrobial agents show an efficient membrane-damaging effect toward negatively charged bacteria, regardless of normal or drug-resistant bacteria [153]. With the development of antimicrobial agents, cationic compounds such as quaternary ammonium (QA), imidazolium, pyridinium, and phosphonium salts are extensively used for the biocidal properties against a broad spectrum of bacteria [154]. However, the antimicrobial efficacy would be highly reduced when cationic polymers are immobilized as antimicrobial coatings, where the diffusion of cationic polymers into cell membranes is highly hindered [155]. As a result, development of novel antimicrobial agents is in constant demand to combat bacterial infections. The combination of photodynamic therapy (PDT) and cationic antimicrobial agents would provide one better way to treat infection cases caused different types of bacteria [156]. To our best knowledge, polycationic synergistic antibacterial agents with integrated QA salt and photosensitizer components are very challenging and have not been reported [157].
Phage and Antibacterial Synergy
Key to any successful drug development is its discovery and subsequent characterization. For phage therapy,
equivalent steps should be taken, including determination of how to combine [158].
Phage isolation is typically done in combination with preliminary host-range characterization, i.e., as regarding enrichment and isolation hosts. This is followed by in vitro characterization in association with further host-range characterization (i.e., involving a larger panel of potential hosts) and bioinformatic (in silico) characterization. Enzybiotic development, if undertaken, typically will follow host-range and in silico characterization. For promising phages, in situ characterization comes next, including animal models for potential human treatments (in vivo characterization), or with other species for non-human treatments. Clinical testing can follow, including treatment of non-human species. Alternatively, phages may be employed for biological control of environments, and both biological control and therapeutic use of phages can be against biofilms. Not only may whole phages be used for therapy or control but so too may enzybiotics. Further development toward successful commercial or public-sector implementation generally must address regulatory requirements.
Phages into multi-phage mixtures known as phage cocktails. The review article in this topic by Weber- Dabrowska et al. discusses the essential steps involved including sources and methods of phage isolation, choice of phage-propagation hosts, methods of characterization, selection criteria for therapeutic purposes, and limitations on phage procurement for therapy. The use of phages as antibacterial therapeutics is especially important for targeting those pathogens for which antibiotic treatment options are limited. On-demand isolation of corresponding phages can be achieved via the enrichment of samples from environmental reservoirs, as explored by Mattila et al. Interestingly, the efficiency of enrichment-based phage isolation from municipal sewage varies considerably, with the best results seen for Pseudomonas Aeruginosa, Salmonella, and the extended spectrum β-lactamase (ESBL) producing Escherichia Coli and Klebsiella Pneumoniae.
The procedure is less efficient for vancomycin-resistant Enterococcus and Acinetobacter baumannii, while isolation of new phages against methicillin-resistant Staphylococcus aureus (MRSA) strains was very difficult. Potentially, the latter may be due to the choice of environmental reservoir used for the anti-MRSA phage isolation since, as Wang et al. show, pig fecal sewage may be a better source for these bacteriophages.
Concluding Remarks
The application of Phage antibacterial therapy is based on a targeted genetic mechanism of bacterial
eradication. I many practical treatments in cases of a targeted resistant bacterial strand, like foot abscesses
our burns, where mainly one resistant bacteria culture caused the infection, success was achieved based on
the unique selectivity of the phages toward the specific bacterium. However, in cases of multiple bacteriabased
infections, combined treatment was found as the most effective treatment, Phage, and broad-band
antimicrobials agents.
Broad-spectrum antibiotics do allow for the treatment of undiagnosed causative agents with some certainty of success. Conversely, even phages with the broadest bacterial spectrums still do not come close to those of broad-spectrum antimicrobials. However, phage narrow host ranges cannot be assumed to exist in nature. It is evident that the clinical application of bacteriophage-based therapy of resistant bacteria-based infections is only in its infancy. More efforts should be allocated to make the combined [159] phage-based antiinfective treatments a real-life saver medical practice.
Novel Approaches to Anti-infective Treatment
In 1982, H. Williams Smith and M. B. Huggins reported on the “Successful Treatment of Experimental
Escherichia coli Infections in Mice Using Phage: its General Superiority over Antibiotics” [160]. Nowadays [161], Phages are promising vehicles for treating Multiple Drug Resistance (MDR), multidrug resistance or multi-resistance is antimicrobial resistance shown by a species of microorganism to multiple antimicrobial drugs. The types most threatening to public health are MDR bacteria that resist multiple antibiotics; other types include MDR viruses, fungi, and parasites (resistant to multiple antifungal, antiviral, and antiparasitic drugs of a wide chemical variety). Recognizing different degrees of MDR, the terms extensively drugresistant (XDR) and pan-drug-resistant (PDR) have been introduced. The definitions were published in 2008 in the journal Clinical Infectious Diseases and are openly accessible [162].

Broad-spectrum antibiotics do allow for the treatment of undiagnosed causative agents with some certainty of success. Conversely, even phages with the broadest bacterial spectrums still do not come close to those of broad-spectrum antimicrobials. However, phage narrow host ranges cannot be assumed to exist in nature. It is evident that the clinical application of bacteriophage-based therapy of resistant bacteria-based infections is only in its infancy. More efforts should be allocated to make the combined phage-based anti-infective treatments a real-life saver medical practice.
Antimicrobial agents take advantage of the differences between animals cells and bacteria (prokaryotes), fungi, or protozoa. The goal is to have highly selective toxicity towards these microbes with minimal or no toxicity in humans. Table 2 shows the basic differences between eukaryotes and prokaryotes.
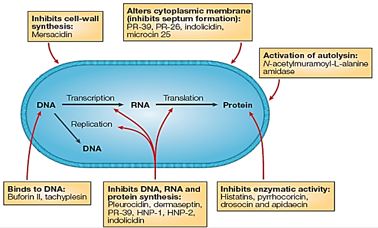
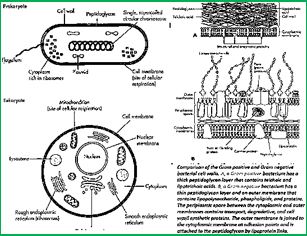
The antimicrobial peptides are promising candidates to model novel antimicrobial synthetic agents. This, due to their promising features centering on the selective eradication of prokaryotes, and the general acceptance of the main route of bioactivity - the corruption of the microbial cell membranes.
However, the unique mechanistic [163] features of these natural, innate immune modulators, did not help when resistant bacteria were investigated [164]. There are several barriers that might impede the development of AMPs as commercial therapeutic agents or restrict their applicability. Among the potential impediments are the high production costs and susceptibility to proteolytic degradation, although AMPs are amenable to extensive chemical modification, which may alleviate some of these obstacles [165]. However, with microbial drug resistance becoming a global public health problem, it has become imperative that new antimicrobials combat the increasing rise in resistance. The development of microbial resistance against AMPs is rare [166]. Nevertheless, microbial pathogens have the capabilities to coordinate countermeasures to circumvent antimicrobial peptide [167] targeting and evade host immune defenses [168].
There is hope in the phage anti-infective therapy, based on the extremes selective manner that phages can react with specific bacteria strands. This, and the genetic mechanism-corruption of the basic genetic molecules of DNA fore instance makes phages as a major component in antiinfective therapy, However, a combination of therapy - antimicrobial agents and specific bacteriophage can demonstrate a way to combat resistant infections.
Bacteriophages are also victims of resistance, microbial defense [169]. Phages are now acknowledged as the most abundant microorganisms on the planet and are also possibly the most diversified. This diversity is mostly driven by their dynamic adaptation when facing selective pressure such as phage resistance mechanisms, which are widespread in bacterial hosts. When infecting bacterial cells, phages face a range of antiviral mechanisms, and they have evolved multiple tactics to avoid, circumvent or subvert these mechanisms to thrive in most environments. In this Review, we highlight the most important antiviral mechanisms of bacteria as well as the counter-attacks used by phages to evade these systems.
Phages are now widely recognized as major ecological contributors in various environments. Throughout the 1950s–1970s, phages were pillars of genetic research and molecular biology. For the past decade, phage research has been going through a renaissance, mainly owing to the prospect of their use in phage therapy or the need to combat them in the food and biotechnology industries. It will be increasingly important to better understand the interactions between phages and their bacterial hosts to fully exploit their antimicrobial potential and to effectively control their populations in bio-industries. For example, it is imperative to go beyond the classical analysis of phage host range and to try to understand why a phage successfully infects one strain but not another.
The Age of Genomics dawned only gradually for bacteriophages. It was 1977 when the genome of phage φX174 was published and 1983 when the “large” genome of phage A hit the streets. More recently, the pace has quickened, so that we now have over 100 complete phage genomes and can expect thousands in a very few years. These sequences have been marvelously informative for the biology of the individual phages. However, but with the advent of high volume sequencing technology, the real excitement for phage biology is that it is now possible to analyze the sequences together and thereby address-for the first time at wholegenome resolution-a set of fundamental biological questions related to populations: What is the structure of the global phage population? What are its dynamics? How do phages evolve? This is Comparative Genomics with a capital “C”.
Streptococcus Pyogenes is a Gram-positive human pathogen with a highly variable clinical symptomatology, ranging from harmless to life-threatening. S. Pyogenes strain SF370 contains eight prophage elements [170]: five prophages have suffered massive deletions, two carry point mutations in key phage genes, and only one prophage is inducible. Similar trends for prophage inactivation exist in other bacteria. A high deletion rate may be a bacterial defense against a high rate of DNA influx by dangerous foreign DNA elements. This hypothetical “cleansing” activity could account for the compact, pseudogene-free nature of most bacterial genomes [171]. Homologous recombination between prophages sharing some DNA homology and residing in the same host can lead to host genome rearrangements and new associations with lysogenic conversion genes in phages.
The recent dramatic expansion of the collection of viral genome sequences, combined with the concerted efforts in evolutionary genomics, translates into a new level of understanding of the origins of the major groups of eukaryotic viruses and the key events in their evolution. We now can delineate both the major general trends in the evolution of eukaryotic viruses and specific scenarios for different virus classes. One of the most striking trends is the distinct composition of the eukaryotic virome compared to the viromes of archaea and bacteria, namely, the high prevalence and enormous diversity of RNA viruses [172].
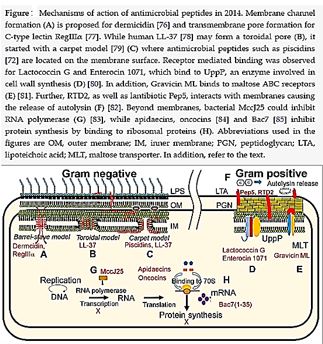
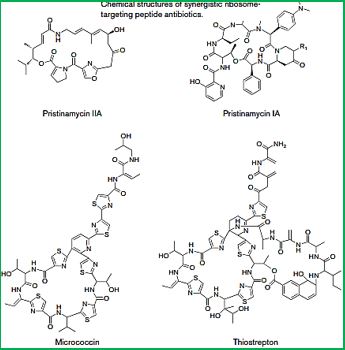
Presently, AMPs represent one of the most promising future strategies for combating infections and microbial drug resistance. This is evident by the increasing number of studies to which these peptides are subjected. As our need for new antimicrobials becomes more pressing, the question remains: can we develop novel drugs based on the design principles of primitive molecules?
Whereas AMP-based therapy relies on the unique mechanisms of bacterial eradication in many regions of the living organisms, Phage therapy targets the genetic materials of the microbes. There are some examples of AMP targeting the same units, however. An AMP must cross the outer envelope, inner envelopes and in Gram-negative microbes has to cross two membranes until the inner cell is achieved. Phages mechanism of action is less demanding.
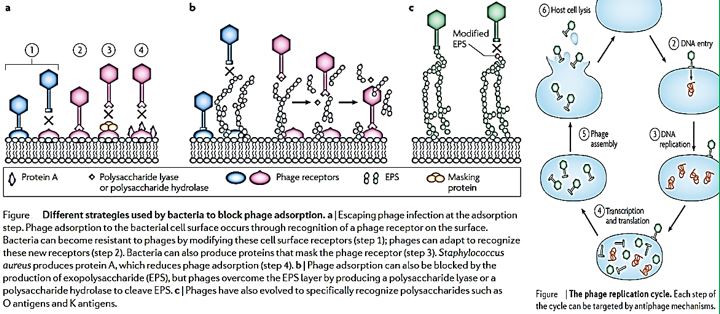
Bacteriophages are not a homogenous group. They are currently classified by their genome (ss versus ds, RNA versus DNA) and their morphology into ten phage families (The Encyclopaedia of Virology, Aca- demic Press, 1999 [173], provides many useful entries for the first orientation on phages). Their diversity is also reflected by the diversity of genome sizes, which ranges from barely 4 kb to up to 600 kb (a mycobacteriophage). 96% of all bacterial viruses are tailed phages (Caudovi- rales, the focus of our writing), which come as Myo-, Sipho- and Podoviridae by tail morphology. Myoviridae have a long contractile tail; Sipho- and Podoviridae have long and short noncontractile tails, respectively. While this classification is quite popu- lar, its evolutionary meaning is far from being clear. Podovirus P22 and Siphovirus A share such a related structural gene map that their taxonomic distinction becomes questionable.
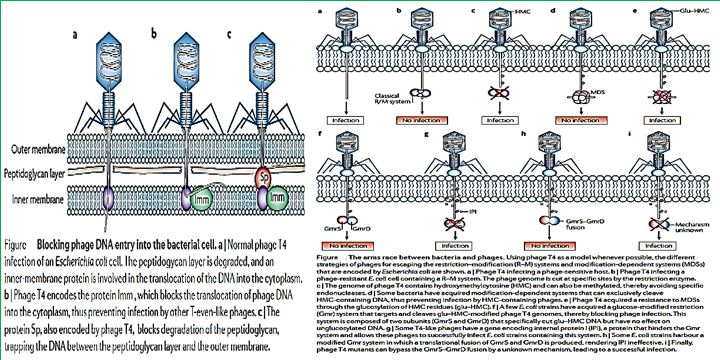
Modulating Tolerance
Infections caused by antibiotic-resistant bacteria continue to challenge physicians. We face growing resistance
among gram-positive and gram-negative pathogens that cause infection in the hospital and in the community.
Recently reported these as the “ESKAPE” pathogens (Enterococcus Faecium, Staphylococcus Aureus, Klebsiella
Pneumonia, Acinetobacter Baumanii, Pseudomonas Aeruginosa, and Enterobacter Species). To emphasize that
they currently cause most US hospital infections and effectively “escape” the effects of antibacterial drugs
[174]. Anti-infectives, the molecular agents (Polymyxins for example), are losing their ability to cure deadly
infections caused by killer bacteria. However, Phage therapy may be a solution to treating infections caused
by antibiotic-resistant bacteria. Researchers have collected bacteriophages to combat antibiotic-resistant
bacterial strains and hope to start clinical phage therapy trials soon.
Multidrug tolerance or antibiotic tolerance is the ability of a disease-causing microorganism to resist being killed by antibiotics or other antimicrobials. It is mechanistically distinct from multidrug resistance: It is not caused by mutant microbes, but rather by microbial cells that exist in a transient, dormant, non-dividing state. Microorganisms that display multidrug tolerance can be bacteria, fungi or parasites.
Anti infective: Anti-infective: An agent that can act against infection, either by inhibiting the spread of an infectious agent or by killing the infectious agent outright.
For more than a half of a century, it becomes more and more evident: there is a transformation in the microbial world, and antibiotic agents are not “magic bullets” anymore. The microbes kill thousands in a new pandemic, the nosocomial illness caused by resistant microbes that prosper in the health facilities, mainly the hospital where people look for remedy.
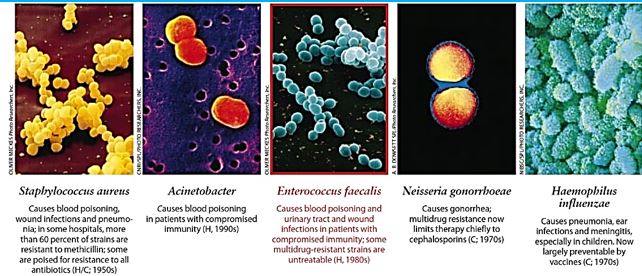
Phage therapy may be a solution to treating infections caused by antibiotic-resistant bacteria. Since 2013, researchers at the University of Helsinki in Finland have collected bacteriophages to combat antibioticresistant bacterial strains, and hope to start clinical phage therapy trials shortly [175].
In the fight against antibiotic-resistant infections, a decades-old approach based on bacteria-slaying viruses called phages has been sidelined by technical hurdles, dogged by regulatory confusion, and largely ignored by drug developers in the West [176]. But two years ago, researchers at the University of California, San Diego (UCSD), used phages to knock out an infection that nearly killed a colleague. Propelled by that success and a handful of others since UCSD is now launching a clinical center to refine phage treatments and help companies bring them to market [177].
Bacteriophages: Natural Enemies of Bacteria
In general, phages start their killing first by recognizing and landing on a bacterium. Each type of phages has
a specific landing pad. The phage then injects its DNA into the bacteria. This DNA copies itself, makes more
of the phage’s shell, and packages the newly made DNA into the new shell. Lastly, the phage produces toxic
chemicals that rupture the bacterial host from inside out, releasing its newly made children to the outside to
infect even more bacteria [178].
Phage therapy or viral phage therapy is the therapeutic use of bacteriophages to treat pathogenic bacterial infections [179]. Phage therapy has many potential applications in human medicine as well as dentistry, veterinary science, and agriculture. If the target host of a phage therapy treatment is not an animal, the term “biocontrol” (as in phage-mediated biocontrol of bacteria) is usually employed, rather than “phage therapy” [180].
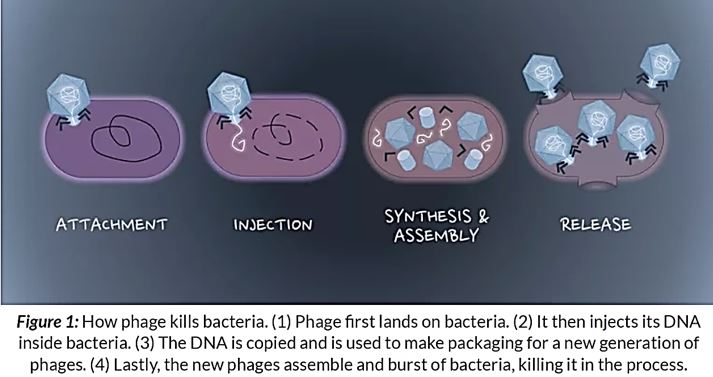
Antibiotic resistance is one of the greatest threats to our societies and health systems, and the number of antibiotic-resistant bacteria is growing at a disquieting rate. The estimated number of people killed by antibiotic resistance each year worldwide is around 700,000, and it is believed it will reach 10 million by 2050 [181].
Bacteriophages, model pathogens, are viruses that ‘eat’ bacteria. They infect and replicate within a bacterium, so they can generally be found wherever you find bacteria, such as in soil. Phage therapy is the therapeutic use of bacteriophages to treat pathogenic bacterial infections. Phage therapy has been used before in humans to treat infections that have either not responded, or become resistant, to antibiotics.
Just recently, the World Health Organization released a report running-out-antibiotics confirming that there is an alarming lack of new antibiotics in development to fight antimicrobial resistance. Scientists are exploring phage therapy as an alternative mechanism for controlling bacterial pathogens [182].
“The silent tsunami, crumbling down the pillars upon which modern medicine is built. Antibiotic resistance that turns ordinary disease-causing bacteria into illnesses that can’t be controlled could bring about the “next pandemic,” Centers for Disease Control and Prevention Director Tom Frieden warned at a National Press.”
Antibiotic-resistant bacterial infections are a major concern for public health. Phage therapy has been proposed as a promising alternative to antibiotics but increasing number of studies suggest that both antimicrobial agents in combination are more effective in controlling pathogenic bacteria than either alone. Health authorities should advocate the use of phages in combination with antibiotics and present the evolutionary basis for our claim.
Hope this find a positive echo in the healthcare community, the 6000 Israeli fatalities [183] in a year due to nosocomial infections could be avoided.
Unlike antibiotics, phages have a very narrow spectrum and often target single species or strains of bacteria. The effect of phage therapy on non-targeted bacteria, however, is largely unstudied. In this study, the impacts of orally administered phage therapy and antibiotic therapy on the composition of the gut microbiome, the complex bacterial ecosystem of the digestive system, were compared [184].
Foreword
Persisters are a small group within a bacterial population that exhibit tolerance to drugs and survive
treatment with antimicrobials. They are phenotypically distinct from antibiotic-resistant bacterial cells,
and drug tolerance in persisters seems to involve the induction of a dormant, metabolically inactive state.
However, although previous studies have identified several mechanisms that promote persistence, it is not
known whether antibiotics effectively enter and accumulate in persister cells. In a recent study, Pu, Zhao, Li
et al. show that in addition to dormancy, enhanced efflux activity contributes to the formation of persister
cells, and suggest that persister cells are latent but active.
Dormant Microbes
Several well-recognized puzzles in microbiology have remained unsolved for decades. These include
latent bacterial infections, unculturable microorganisms, persister cells and biofilm multidrug tolerance.
Accumulating evidence suggests that these seemingly disparate phenomena result from the ability of bacteria
to enter into a dormant (non-dividing) state [185].
Bacteriophages were first used successfully to treat bacterial infections a decade before penicillin was discovered. However, the excitement that greeted those initial successes was short-lived, as a lack of understanding of basic phage biology subsequently led to a catalog of clinical failures. Therefore, bacteriophage therapy was largely abandoned in the West in favor of the newly emerging antibiotics.
Now, as the problem of antibiotic resistance becomes ever acuter, several scientists and clinicians are looking again at bacteriophages as a therapeutic option in the treatment of bacterial infections. The chances of success second time round would appear to be much better given our current extensive knowledge of bacteriophage biology following their important role in underpinning the advances in molecular biology. We also have available to us the experience of nearly 80 years of clinical usage in the countries of the former Soviet Union and Eastern Europe as well as a political climate that encourages sharing of that knowledge. This paper outlines those features of bacteriophages that contribute to their utility in therapy and explores the potential for their re-introduction into Western medicine. An abundance of clinical evidence is available in the Soviet literature but much of this is technically flawed, and a more realistic appraisal of the clinical value of phages can be obtained from animal studies conducted in the West. As interest in bacteriophages increases, some companies throughout the world have begun investing in phage technology and this has led to novel approaches to therapy, some of which will be discussed [186].
A dormant cell has a global slowdown of metabolic processes and does not divide.
Dormancy is a period in a organism’s life cycle when growth, development, and (in animals) physical activity are temporarily stopped. This minimizes metabolic activity and therefore helps an organism to conserve energy. Dormancy tends to be closely associated with environmental conditions. Organisms can synchronize entry to a dormant phase with their environment through predictive or consequential means. Predictive dormancy occurs when an organism enters a dormant phase before the onset of adverse conditions. For example, photoperiod and decreasing temperature are used by many plants to predict the onset of winter. Consequential dormancy occurs when organisms enter a dormant phase after adverse conditions have arisen. This is commonly found in areas with an unpredictable climate. While very sudden changes in conditions may lead to a high mortality rate among animals relying on consequential dormancy, its use can be advantageous, as organisms remain active longer and are therefore able to make greater use of available resources.
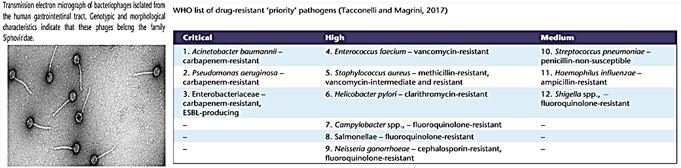
That Resistant, Persisted, Dormant and other microbes that are not eradicated by the currently applied anti-infective [187] agent, are the candidates for phage antimicrobial therapy. Unfortunately, phage therapy is more cumbersome that antibiotic treatment. No tablets or injections are commonly applied during hospitalization is needed.
Since their discovery in 1915, bacteriophages have been used to treat bacterial infections in animals and humans because of their unique ability to infect their specific bacterial hosts without affecting other bacterial populations [188]. Bacteriophage (phage) therapy has encountered both enthusiasm and skepticism in the past century. New antimicrobial strategies against lethal pathogens are now a top priority for the World Health Organization, and although the compassionate use of phages recently met with significant success, regulated clinical interventions seem unlikely shortly. The hundredth anniversary of their discovery seems an appropriate time for a revival of phage therapy, particularly as the dilemma of antibiotic resistance grows. Phages are ubiquitous in the environment, on our food and in and on our bodies. Their influence on human health is currently being evaluated, and in this mini-review, we examine data from recent metagenomic studies that propose a role for phages in the structure of the microbiome and in health and disease. We assess evidence for phages as vehicles for gene transfer in the context of antibiotic resistance and discuss challenges and opportunities along the critical path from phage discovery to a patient-focused pharmaceutical intervention [189].
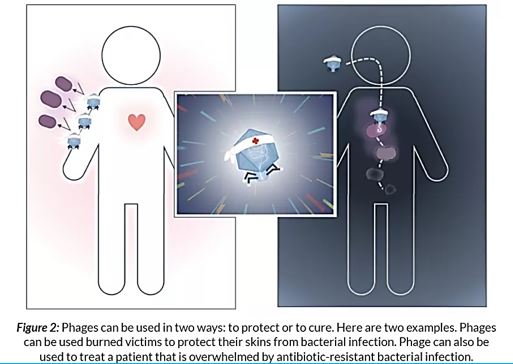
However, more experience and Pharmaceutical technology are needed to bring this Phage-based therapy to the applied level of application known in the antibiotic treatment practice.
Metagenomics is a powerful tool for deciphering the diversity of microbes in the human body. We now predict that the human microbiota, in particular, the gut microbiota, plays a major role in health and well-being. We are in a period of discovery about the role of phages in the overall interplay between host and microbiome and, as we learn more from metagenomic and pharmacological studies, we will be able to identify candidate targets of diagnostic pharmaceutical and therapeutic significance. Although phage therapy against.
Pathogenic infections of humans have a century-long history in Eastern Europe and former Soviet countries, approved use in Western society still requires several clinical, manufacturing and regulatory hurdles to be overcome. Ongoing dialogue between scientists, health care professionals, pharmaceutical companies, regulatory authorities and policy-makers is imperative to implement phage therapy in clinical practice. In light of the WHO’s urgency for novel agents to target the world’s most life-threatening superbugs, the question is not whether we should pursue phage therapy as a solution, but whether we can risk not pursuing it [17].
The misuse of antibiotics has reduced their efficacy in controlling pathogens and has led to an increase in the number of antibiotic-resistant bacteria. As an alternative to antibiotics, bacteriophages have become a topic of interest with the emergence of multidrug-resistant bacteria, which are a threat to public health. Recent studies have indicated that bacteriophages can be used indirectly to detect pathogenic bacteria or directly as biocontrol agents. Moreover, they can be used to develop new molecules for clinical applications, vaccine production, drug design, and in the nanomedicine field via phage display.
The Use of phages in as anti-infective in therapy can be extremely successful based on the selectivity of the bacteriophages toward bacteria. Also, the mechanism of action directed at the genetic materials in the nuclei of the microbial cells is different from the cell wall disruption characteristic to most antibacterial agents, mainly antimicrobial peptides and their surrogates. However, Hospital-acquired infections are; like postsurgical infections, are characterized by their multi-infectors blend.
We can see more than 5 infectors in the post-surgical infection in the Ethiopian clinic. Here we can find many sorts of infectors, Gram-negative like Klebsiella spp. and Gram-positive S. Aureus, treatment of such wounds will apply a mixture of phages, whereas today’s treatment can apply one agent for all microbes [190].
Although use of biologics in infection control is still in its infancy, its potential for combating multi-drug resistance cannot be ignored [191]. Small molecules will always have a place in infection control; however, effective candidate development may be more intelligently pursued based on biological inspiration. Drug development tools necessary to overcome the pitfalls of biologics, including limited in vitro stability, restricted delivery options, limited high-throughput, discovery and development screening tools, imperfect pharmacokinetics and relatively unexplored pharmacodynamics, are not as advanced as those available for small molecule development. Fortunately, limitless potential exists in the combinations of biologics, biologically inspired molecules, and drug delivery technologies. Thus, a paradigm shifts akin to that necessary for complex viruses and cancers is essential to design a targeted attack that eradicates susceptible microbes, contains and squelches microbial resistance, and protects the host flora from the therapeutic. Such a strategy may arise from combinations of traditional antibiotics, new adjuvants, and sustainable localized delivery approaches. Using advanced bioinformatics to predict successful combination delivery and novel targets may pose significant advantages over current costly development models that often fail in late phase trials [192].
Bibliography


Hi!
We're here to answer your questions!
Send us a message via Whatsapp, and we'll reply the moment we're available!