Biography
Interests
Stavroula Koulocheri, A.
Department of Biological Chemistry, Medical School, National and Kapodistrian University of Athens, NKUA, Greece
*Correspondence to: Dr. Stavroula Koulocheri, A., Department of Biological Chemistry, Medical School, National and Kapodistrian University of Athens, NKUA, Greece.
Copyright © 2019 Dr. Stavroula Koulocheri, A. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
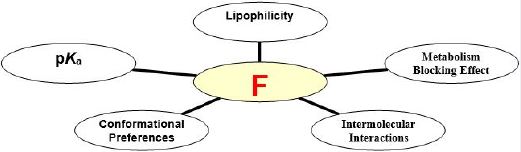
The highly electronegative and unique fluorine incorporated to organic compounds can play an essential role in medicinal chemistry. The rational introduction of fluorine has the potential to modulate pKa, potency, membrane permeability and influence conformation, protein binding and pharmacokinetic properties. There are many applications of fluorine substitution in drug discovery and are progressively increasing in parallel with the development of new synthetic strategies and methodologies.
In this review the exploration of the interesting characteristics of the C-F bond in organofluorine compounds is described and some examples of marketed fluorinated medicines are shown. The many advantages of fluorine in candidate drugs constitute a great challenge for further optimisation and future development of drugs with high potency and efficacy.
Introduction
Fluorine’s unique physicochemical properties make it a key element for incorporation into bioactive
compounds [1]. Its presence in a drug can modulate a number of characteristics that affect ADME
(Absorption, Distribution, Metabolism, Excretion). Over the last two decades, fluorine substitution has
become one of the essential structural characteristics in modern medicines [2,3].
The use of fluorinated drugs to a wide range of therapeutic areas shows the extensive role they play in modern drug development and application. Currently, many fluorinated compounds are synthesised routinely and are widely used in the treatment of diseases as anti-cancer drugs, antidepressants, anti-inflammatory agents. To provide only a few examples, the cholesterol-lowering drug atorvastatin (Lipitor), the COX-2-specific nonsteroidal antiinflammatory drug celecoxib (Celebrex), the antidepressant drug fluoxetine (Prozac), the antidiabetic drug sitagliptin (Januvia) and the antimalarial drug ciprofloxacin (Cipro) [4].
Two known drugs discovered in 1950s which contained fluorine atoms, are 9α- fluorohydrocortisone (an anti-inflammatory drug) [5] and 5 - fluorouracil (an anticancer drug) [6].
Since then, the incorporation of fluorine into pharmaceutical drugs in order to increase their pharmacological action has become almost a common procedure. Approximately 20% of currently available drugs are fluorinated with fluoroaryl, simple fluoroalkyl and aromatic trifluoromethyl groups present in 80% of compounds and the number of all new pharmaceuticals with fluorinated sub-units is increasing [7].
In 2006, a bestselling drug was Lipitor® (atorvastatin calcium) for the treatment of hypercholesterolaemia by Pfizer/Astellas (Figure 1).
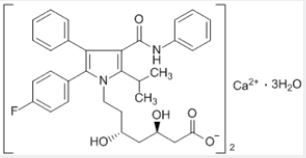
The advantages of fluorine-containing drugs promote the research on the synthesis of organofluorine compounds in medicinal chemistry for drug discovery. In most cases, 1 - 3 fluorines are incorporated in place of hydroxyl groups or hydrogen atoms (usually CHF, CF2, CF3 groups). There is a continuous need for the development of improved and efficient synthetic methods for biological organofluorine compounds [9].
In this short review some notable properties of fluoro-organic molecules, important in medicinal chemistry are presented from the literature. Fluorination in organic molecules creates important features and significant pharmacological properties. This review describes the physicochemical characteristics of the element and its influence and relation on features like potency, lipophilicity, metabolic stability and bioavailablility. Fluoroorganic molecules and the effects of fluorine on drugs have been comprehensively reviewed in recent years, and several excellent reviews covering various areas of fluorine in pharmaceuticals have been published [10- 13], so only a brief overview is presented below.
In this review some of the unique properties of fluorine in medicinal chemistry will be discussed and some selected examples of marketed fluorinated medicines will be mentioned in the last section of the article (Figure 6). Synthetic methods, mechanisms and strategies for introduction of fluorine into drug leads will not be discussed.
Effects of Fluorine in Organic Compounds
It is known that the van der Waals radius of fluorine is about 20% larger than that of hydrogen. In particular,
fluorine’s small van der Waals radius, coupled with its electronegativity (4 on the Pauling scale, the most
electronegative element) fluorine can be incorporated into a drug molecule with little steric effect but this
creates great considerable electronic consequences. The C - F bond length in CH3F is a little longer than the
methane’ s C - H bond [14]. The replacement of C-H bond with C-F results in “similarity” of the natural
substrate and its analogue. Thus enzymes and microorganisms are not being able to discern the difference. This
is called mimic effect [10]. The use of fluorine substitution has found considerable use in blocking oxidative
metabolism at both aromatic and aliphatic labile sites of drug-candidates. The replacement of hydrogen with
fluorine is a very effective way to slow down considerably the oxidative metabolic pathway of a given drug
by cytochrome P450 enzymes. Hydrolytic metabolism can also be noticeably affected by fluorination. In this
case the electron-withdrawing effect of fluorine exerts a key role in changing reaction rates and the stability
of the intermediates. The C-F bond is very strong, a property widely used by medicinal chemists for blocking
the metabolism of drug molecules. This procedure has been used not only to decrease the rate of metabolic
oxidation but has also been used to prevent the generation of reactive and toxic molecules. Aromatic and
aliphatic oxidations are similar but there are differences in oxidation mechanisms. In aromatic oxidation, the
initiation step begins with ring epoxidation. In aliphatic oxidation the first step is hydrogen atom abstraction
to give a radical species that is subsequently oxidized to give an alcohol. Thus given the fact that H3C - F
bond is stronger than that of H3C - H, metabolic oxidation reactions via hydroxylation of C - H bonds, by
the cytochrome P - 450 of enzymes, can be blocked because of the replacement of a specific C - H bond with
a C - F bond. This is known as a block effect. A classic example of this is the cholesterol transport inhibitor
Ezetimibe (Zetia™, Merck/Schering Plough Pharmaceuticals) (Figure 2), in which labile sites in oxidation
attacks, resulting in reduced efficacy, were fluorinated, showing that the replacement of an oxidizable for
example C-H group by a C-F group increases metabolic stability of the molecule [15]. Also the oxidation of
metabolically labile groups on aromatic rings like methoxy groups can be prevented by fluorine.
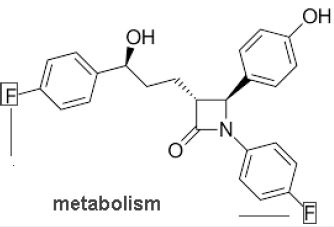
Consequently, the purpose of fluorine incorporation into drug molecules in place of hydrogen is often to slow or prevent metabolic attack, by increasing resistance to cytochrome P450 (CYP)-mediated oxidation. So there is a longer duration of the pharmacological effects of the drug. Due to the increased metabolic stability of the fluorinated drugs, their bioavailability is increased. Thus, there is the potentiality of introduction fluorine(s) into labile metabolism site(s) leading to modification of the biological activity of compounds. In addition to these effects, the introduction of the electronegative fluorines results in negative inductive effect (-I) on neighbouring functional groups. Carboxyl and amino groups have smaller pKa values and the Lewis basicity is decreasing altering oxidation process [10]. Generally, the rational introduction of fluorine into a molecule can influence pKa, potency, membrane permeability, metabolism, pharmacokinetic behavior and conformational preferences.
The absorption and distribution of a medicinal molecule in vivo depend on the relation of lipophilicity,
hydrophilicity and ionization. Fluorine substitution in drug candidates, by means of its exerting increased
lipophilicity to the molecules, helps in the tissue absorption and distribution. It is generally known that
incorporation of fluorine or fluorinated groups in organic compounds leads to an increase in lipophilicity
despite the C-F bond’s high polarity, especially in aromatic compounds. In addition, because of the increased
metabolic stability of the fluorinated drugs, their bioavailability is increased. The lipophilicity of the drugs is
measured as the logarithmic partition coefficient, logP, between n-octanol and water; the higher the logP,
the higher is the lipophilicity of the compound.
When fluorine is incorporated closer to the amino groups, the amine basicity is decreased due to the strong -I inductive effect of the fluorine, so that the amines exist mostly in the neutral form and the corresponding log P values and their lipophilicities are higher. Increased lipophilicity and change of amine pKa results in greater in blood - brain barrier (BBB) permeability [16]. This introduction is associated with the term polar hydrophobicity [17]. The term polar hydrophobic is finding increasing use to recognise that the electron withdrawing power of the C-F bond reduces the polarisability of molecules and increases their hydrophobicity [18]. Fluorobenzene is more lipophilic than benzene. Similarly, CF3 - Ph (phenyl) is much more lipophilic than CH3- Ph. In case where there is the CF3 -group, the strong negative inductive effect (-I) near polar groups in these compounds as amine, alcohol, because of the -I effect, there is a decrease in electron density of the polar groups and decreased participation in hydrogen-bonding. The consequent increase in hydrophobicity means increased lipophilicity. Many drugs have fluorophenyl and trifluoromethyl phenyl groups in their structures and the increased lipophilicity may enhance partitioning into cell membranes, raising the effective intracellular concentration. The effect of fluorine substitution on lipophilicity in aliphatic compounds is more complex. Changes in lipophilicity are shown by measuring octanol–water distribution coefficients (logD), and are related to the presence of charged groups in molecules. pKa can influence logD significantly, and negative inductive effects such as fluorine’s can have additional effects on logD. The introduction of fluorine into aliphatic compounds results in decrease in lipophilicity as a direct consequence of the relatively polar character of monofluoro-and trifluoro - methylalkanes with the strong C-F and C-CF3 bond dipoles, but the examples are limited [19]. Regarding straight - chain alcohols with a terminal CF3, these are more lipophilic than nonfluorinated counterparts [20]. Trifluoroethanol is more lipophilic than ethanol because of the decrease in the basicity of the hydroxyl group by the strong negative inductive effect of electron CF3 group. However the σ-inductive effect is decreased with distance (after carbon four) and there is a reversal of lipophilicity.
A recent study looking at the sequential impact of terminal fluorination of alkyl chains indicates that the order of decrease in log P/D (partition coefficient: log P, distribution coefficient: log D) follows the sequence CH3<CF3<<CHF2≤CH2F and is related to the magnitude of the dipole in each case [21,11].
The more lipophilic the drug candidate, greater effective concentration of the drug exists at the enzyme/receptor active sites. It is known that the active site regions of enzymes have relatively decreased polarity, thus the lipophilic molecules can improve the binding to the hydrophobic binding pocket of the target protein, binding with more affinity. So, certain fluorinated ligands-drugs at the active sites, can function efficiently in lower concentrations.
Absorption, distribution, metabolism, excretion and toxicity (ADMET) are significantly affected by the
charge state of molecules under various pH conditions. Consideration of pKa values in conjunction with
other molecular characteristics is of great significance. The behaviour of drugs depends on their acid-base
character which affects their biopharmaceutical characteristics and is really of great importance. Essentially,
the acid-base profile of a molecule has a direct effect on the lipophilicity as governed by the ionisation
constants, pKas of key functional groups.
Modulation of pKa is often an important objective in drug development, affecting molecular interactions or physicochemical features. Also fluorine or fluorine moiety of organic compounds affects binding affinities, receptor-ligand or enzyme-inhibitor binding and biological responses.
It is known that fluorine is the most electronegative element with strong withdrawal σ-inductive effect (-I). This influences the physicochemical properties of the molecules containing fluorine groups. Because of the powerful negative inductive effect of a fluorine substitution and its moderate impact on size, shape, fluorine substitution is a good choice for pKa modulation of acidic and basic groups. Thus F substitution is a powerful tool for pKa modulation. The introduction of fluorine into organic compounds causes considerable changes in pKa values of carboxylic acids, alcohols, and protonated amines. The presence of the electronegative fluorine creates strong decreases in pKas. Thus, fluoreacetic acid has smaller pKa than acetic acid. The negative inductive effect through σ bonds is disappearing with the distance.The introduction of two or three fluorines increase more the acidity. Introduction of CF3 groups to alkanoles increases considerably the acidity of the alcohols. Also perfluorination of benzoic acid increases the acidity [22].
On the contrary, the introduction of fluorine(s) to aliphatic amines decreases their basicity which means higher bioavailability, and lipophilicity which is the proportion of drug reaching the circulatory system [23].
The inductive effect of fluorine is sufficient to render an amino- group nonbasic and sufficiently polarized to act as a hydrogen bond donor. Concerning the modulation of pKa of bases in drug molecules, the pKa of fluorinated alkyl amines are reduced through fluorination, with the impact diminishing with distance from amino group because of the -I effect of fluorine.
The inductive effects of fluorine modulate the properties of some enzyme inhibitors and contribute to drug design and development in terms of changes in pKas, ionization states and binding affinities of inhibitors with the corresponding enzymes.
For example, in the heteroaryl sulphonamides which are carbonic anhydrase inhibitors, it has been shown that the relationship between potency and pKa in halogenated analogues of methanesulphonamide was linear [24]. The trifluoromethanesulphonamide analogue was the more acidic and more potent than the methanesulphonamide.
It is worth to note the impact of fluorination in reducing efflux potential through pKa modulation of drugcandidates.
Given the fact that acid-base profiles and pKa values are very important affecting all aspects of drug discovery, it is useful to analyse continually the acid-base properties of drug-candidates enhancing our knowledge in this field and improving drug discovery processes.
There are conformational effects of fluorine on organic molecules and are caused by the features of the
C-F bond [1,25]. The molecular orbitals of fluorine are energically very low - lying because fluorine is very
electronegative. A C - F bond can readily accept electrons to its vacant σ * C - F antibonding orbital from a
neighbour electron - donating orbital, or oxygen and nitrogen lone pairs [1], given the fact that the electron
- occupied σ bonding C - F orbital is reluctant to donate electrons. This allows more electron-rich bonds
antiperiplanar to the C-F bond to donate electron density to the σ*C-F antibonding orbital. This behavior
can have effects to the three - dimensional shape of a molecule in a particular mode. A representative example
is 1,2 - difluoroethane with the known gauche and anti-possible conformations. The molecule adopts the gauche conformation because is more stable [26] and this is called gauche effect, stabilization through
the donation of electrons from the neighbouring σ C - H bonding orbital to the lower - lying vacant σ
* antibonding C - F orbital. Charge-dipole interactions can lead to considerable preference for gauche
interactions between a fluorine substituent and a protonated amine in open chain molecules. The gauche
conformation is favoured by up to 5.8kcal/mol. This gauche effect is useful for exploring the conformational
preferences of various ligands. Generally, in aliphatic systems, a combination of these effects gives a strong
preference for adjacent functional group to align gauche to fluorine. This influence is great and finds application
in drug design.
Also negative hyperconjugation of fluorine [27], the same electron donation mode as that of the gauche effect, that is, interaction of an electron - rich bond with the lower - lying vacant orbital of a polarized neighboring C - F bond that is σ * antibonding C - F orbital, has been clearly observed in X - ray structures.
So, there is the potential for the fluorine gauche effect - hyperconjugation to influence activity of fluorinated analogues and this interaction has been mentioned in order to make clear the differences in potency between fluorinated analogues of the HIV protease inhibitor indinavir [28,11].
Sometimes, it is worth to mention that differences in activity have been connected to the various conformational preferences of drug-candidates.
Fluorine Interactions in Proteins
There is a great interest for the role that organofluorine may play in binding interactions with proteins with
changes in binding potency of the fluorinated compound versus its hydrocarbon parent. Because of the
unusual properties of fluorine and the increases in lipophilicity, it is expected that fluorine interactions with
proteins, could give rise a particular pattern of binding interactions.
X-ray crystal structures from the Royal Society of Chemistry protein database, of fluorine-containing compounds bound to their receptors, show some preferred environments [29].
Attention has shifted on intermolecular interactions of organofluorine, such as H-bonding and dipolar interactions. Particular organofluorine interactions with protein residues can be used to considerably enhance protein-ligand binding affinity and selectivity.
From Lewis structure of fluorine, there are three sets of lone - pair electrons for sharing with electron -
deficient atoms in an intramolecular or intermolecularl mode, particularly with a relatively acidic hydrogen.
Perfluoroalkyl groups showing great negative inductive effect (-I), increase the acidity of neighbouring
functional groups such as alcohol, amine, amide and carboxylic acid. Thus, this means that the benzylic
hydroxyl group should form a hydrogen bond with a CF3 group [30]. Similar hydrogen bonding pattern
has been between fluorine and amine or amide hydrogen. The NH · · · F hydrogen bond is suitable with the amide hydrogen because of its acidity compared with that of an amine. It has been shown that fluorinated
benzenes, form intermolecular hydrogen bonds in the solid state. It must be emphasized that due to the high
electronegativity of the fluorine, the hydrogen bonding ability of fluorine is significantly lower than that of
the oxygen or nitrogen atoms. In a C-F bond the fluorine lone pairs are held by the nucleus (electronegativity)
and the adjacent partially charged (δ+C) carbon, so they are not easily polarised and are not good hydrogen
bonding acceptors. Thus, the interaction of the fluorine with its environment is mainly through electrostatic
interactions [1].
The great strength of the C-F bond can be ascribed to significant electrostatic attraction between Fδ- and
Cδ+ rather than the more classical electron sharing of a covalent bond. The strong ionic character of the C-F
bond creates a large dipole moment that can also influence the conformational behaviour of organofluorine
molecules [31]. In the case of α-fluoroamides, the orientation of C-F bond is antiperiplanar to the amide,
such that the C-F and amide dipoles are in opposite direction and this is called α-fluoroamide effect [32].
The strongest intermolecular interactions of the C-F bond is expected in charge-dipole interactions between C-F and a formally charged heteroatom [33]. Thus this charge-dipole interaction of the C-F bond has a potential as a means for examining the binding mode of protonated amines on target proteins. Usually, in fluorine interactions with proteins, fluorine is not a good H-bond acceptor. The C-F dipole preferentially undergoes multipolar interactions (these are described in the next section) to backbone amide NHs, the carbon of backbone carbonyl groups, α-carbon CHs and guanidinium groups [29,34].
Fluorine to hydrogen interactions-contacts are described in the literature as either hydrogen bonding or more often multipolar interaction in nature, due to the ambiguity around fluorine and H-bonding.
It has recently been shown that polar C - F bond - protein interactions play an essential role in the stabilization
of fluorinated drugs and their target proteins [34]. There are many examples of interaction between the polar
C - F bond and proteins through database mining. Fluorine, due to its highest electronegativity, is a weak
H-bond acceptor.
However, protein–ligand X-ray structures indicate many cases in which fluorine engages in multipolar, H-bond type interactions; that is, C-F…H-N, C-F…C=O, C-F…-Cα interactions, involving protein backbones or side chains, in many X-ray structures of the enzyme- substrate complexes. These interactions are a C - F bond and polar functional groups such as carbonyl and guanidinium ion moieties in the protein side chains, of the type C - F · · · C = O and C - F · · · C(NH2)( = NH). From the protein crystallographic databases, it has been shown that a C - F bond is as a poor hydrogen bond acceptor. However instead of hydrogen bonding, (because of poor lone pair donating power) a C - F bond forms dipolar interactions with a large number of dipole- bonds in the mode as C - F · · · X(H) (X = O, N, S) and C - F · · · C α- H (C α carbon of the amino acid), in which the F · · · H - X distance is well beyond hydrogen - bonding contact distance [34].
It is worth noting that a thrombin inhibitor is more potent than the nonfluorinated one and the X - ray crystal structure of the inhibitor - enzyme complexes showed notable conformational differences between the two inhibitors. This conformational change is caused by the dipolar C - F · · · N(H)(Gly216).
A large number of examples exist for the orthogonal dipolar C - F · · · C = O and double C - F · · · (C = O)2 interaction in inhibitor - enzyme complexes. It is worthy of mention that the C - F bond and the electrophilic carbonyl group are positioned in a nearly orthogonal pattern.
A polar interaction exists between a C - F and the guanidinium carbon of the Arg residue in X - ray crystal structure of atorvastatin - HMG - CoA reductase (HMG - CoA : 3 - hydroxy - 3 - methylglutaryl -coenzyme A). Atorvastatin is a best - selling cholesterol - lowering drug, and binds to HMG - CoA reductase tightly (IC 50 = 8nM). The X - ray crystal structure of atorvastatin – HMG - CoA reductase [35] shows a strong C - F · · · C(NH2 )( = NH) polar interaction between the 4 - fluorophenyl moiety and the Arg590 residue of the enzyme (Figure 3) [34].
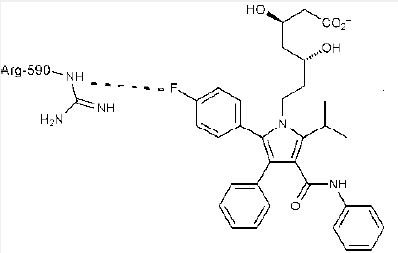
Many similar polar interactions between a C - F bond in a ligand and the guanidinium ion of an Arg residue in proteins have been found in PDB. On the contrary, no linear C - F · · · H - N interactions are observed, showing the poor hydrogen -bond accepting ability of a C - F bond. Instead of hydrogen bonding, a C - F bond is found in an orientation parallel or orthogonal to the plane of the guanidinium ion showing a delocalized positive charge. These examples prove the fluorophilic character of the guanidinium side - chain of an Arg residue [10]. Protein backbone amide carbonyl groups, positively charged side chains of arginine, glutamine, and guanidinium, serve as fluorophilic environments for a ligand at the enzyme active sites. Also, acidity (or basicity) of the key functional groups can be fine-tuned by the fluorines (through the electronwithdrawing inductive effect (-I) of fluorine) in order to modulate the enzyme-substrate(ligand) binding affinities. C-F…C=O interactions can play an important role in protein-ligand interactions and can lead to significantly enhanced binding affinities [36].
It is recognized that fluorophilic environments possibly exist in proteins. It is worth of note that NMRshielded and NMR-deshielded fluorines have different binding site preferences [37,38]. Shielded fluorines are observed preferentially close to hydrogen bond donors in the protein, resulting in forming possibly intermolecular H-bond. Deshielded fluorines are possibly found near to hydrophobic side chains, the carbonyl carbon of protein backbones and the sulphur of methionines.
Isosteres
It is known that natural and synthetic peptides exhibit biological activities, which can be used as
pharmaceutical drugs. However, the amide linkages are easily deactivated through cleavage by hydrolytic
enzymes. Consequently, it is very useful if an essential amide bond is replaced with noncleavable bond,
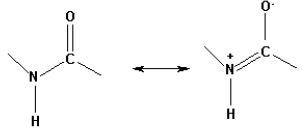
maintaining the amide functionality. An amide linkage has an imidate - like zwitterionic resonance structure.
The contribution of this resonance structure is significant and free rotation around the C - N bond is partially
restricted because of the double - bond character of the C - N bond (Figure 4). Accordingly, it might be
possible to replace an amide linkage with its isostere (isostere is a molecule having the same number of
atoms as well as valence electrons) a peptide isostere (Figure 5) [39].
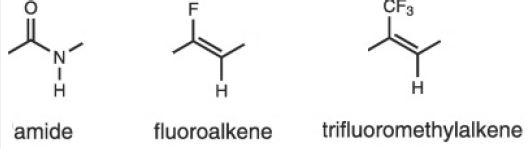
A trifluoromethyl group is a better electrostatic mimic of the carbonyl oxygen of an amide than the fluorine of fluoroalkene based on the fact that the dipole moment of trifluoromethylalkene (2.3D) is closer to that of an amide (3.6D) [40].
A fluoroolefin isostere was introduced to a physiologically important neuropeptide, Substance P [41]. Fluoroethene and trifluoroethene peptide isosteres were used as nonhydrolyzable amide substitutes. It is worth of mentioning trifluoroethylamines that are considered as highly promising peptide isosteres. Also, it has been demonstrated that the CF3 group of the trifluoroethylamine isostere is able to function as the carbonyl group of an amide to offer a metabolically stable, nonbasic amine that has excellent hydrogen - bonding ability because of the strong negative inductive effect of the CF3 group [42]. These features of the trifluoroethylamine isostere have been used and successfully applied to the optimization of cathepsin K inhibitors, which are promising antiresorptive agents for treatment of osteoporosis [42a].
Difluoromethylene as Isopolar Mimic of the Oxygen Component of P - O - C Linkage
The difluoromethylene moiety has been used as an electronic and steric mimic of labile oxygen atoms in
phosphate esters (R-CF2-PO3
2- vs R-O-PO3
2-). Difluoromethylene has been shown as an isopolar mimic
of the oxygen component of P - O - C or P - O - P linkage in phosphates, which can be used to generate
nonhydrolyzable phosphate analogues of nucleotides, enzyme substrates, and enzyme inhibitors [43].
For example, a difluoromethylene linkage was successfully introduced to a protein - tyrosine phosphatase (PTP) inhibitor of insulin receptor dephosphorylation [43a].
Use of Difluoromethyl and Trifluoromethyl Ketones as Transition State Analogues in
Enzyme Inhibition
Tetrahedral transition state analogues of ester and amide substrates are known to function as effective enzyme
inhibitors of hydrolytic enzymes such as serine and aspartyl proteases as well as metalloproteinases [44].
Although ketals of alkyl or arylketones are usually not stable, those of difluroalkyl or trifluoromethyl ketones have considerable stability [45]. Thus substrate analogues, containing difluoroalkyl or trifluoromethyl ketone part in appropriate positions, are considered as effective transition state inhibitors of hydrolytic enzymes [46].
The SF5 group offers interesting properties and an unusual geometry that have potential use in drug design.
However, the applications are rare because of the synthetic difficulties. The lipophilicity and electron-withdrawing
inductive effects of the SF5 moiety are higher than those of CF3. The shape of SF5 is octahedral and
has larger size than the tetrahedral CF3.
Although there is not yet any experience concerning drug design, alkyl SF5 derivatives have shown to affect conformational preferences due to steric and stereoelectronic effects [47].
Fluorine as a Key Element in Drugs-Examples
As mentioned in the introduction, there are many marketed drugs containing one or more fluorine atoms.
Examples of commercially important fluorinated pharmaceuticals (Figure 6):
Conclusion
Fluorinated drugs synthesized with modern medicinal techniques have led to a large number of highly effective
drugs. F introduction to organic compounds can influence potency, selectivity, absorption and metabolism.
Besides the blocking effect in metabolism, fluorine is also introduced to modulate the physicochemical
properties and to enhance binding affinity through specific interactions of fluorine-containing ligand with
the target protein. It is becoming clear that F can enhance binding efficacy and selectivity in pharmaceuticals.
Great resources are being invested in pharmaceutical industry for studies of various structure-activity
relationships of fluorine substitution. Fluorine could be involved in a more rational approach to drug design
for maximising activity. Modern fluorine-organic chemistry has provided new significant synthetic routes
for the specific introduction of fluorine into organic molecules. Organofluorine molecules as drug candidates
have enhanced our understanding of the molecular mechanisms of disease states and are continuously
contributing to the development of drug discovery. It is hoped that F substitution will continually offer
interesting new opportunities in medicinal chemistry.
Bibliography


Hi!
We're here to answer your questions!
Send us a message via Whatsapp, and we'll reply the moment we're available!