Biography
Interests
Rajneesh Verma3, Shashi Paul1, Balamuralidhara, V.1* & Manoj Bansode2
1Department of Pharmaceutics, JSS College of Pharmacy, Sri Shivarathreeshwara Nagara, Mysuru
2Saiseva Biotech Pvt Ltd, CureCellsTM Cord Blood Bank, Kant Helix, Bhoir colony, Chinchwad, Pune
3Stem Cell 21 Co. Ltd 2nd & 7th fl, Urbis bld, Aetas Residence, Soi Ruamrudee, Bangkok, Thailand
*Correspondence to: Dr. Balamuralidhara, V., Department of Pharmaceutics, JSS College of Pharmacy, Sri Shivarathreeshwara Nagara, Mysuru.
Copyright © 2019 Dr. Balamuralidhara, V., et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
Development of wide range of medical devices across the world resulted in the urge of unique challenges like safety concerns coupled with the diversity of products. Therefore, regulators and governments count on standards to help develop better regulation to set standards so as to develop an effective and efficient regulatory scheme. Different regulatory bodies have set regulatory requirements for manufacturing of the medical devices. Medical Device Regulations, 2017 provides detailed information about procedure for filing of application of Medical Devices to the CDSCO for its approval in India with respect to each category of medical devices which are defined on the basis of the risk involved and different parameters like intended use, type of use etc. These regulations provide the elaborately defined requirements of facilities, quality management system to be followed, regulatory timelines, regulatory requirements and approval procedure, area requirements with respect to environment condition, clinical investigation requirements for New Medical Device/investigational Medical Device, labeling for manufacturing of the Medical device in India. These Regulations are framed by the Ministry of Health And Family Welfare under the and Cosmetics Act, 1940 and Rules, 1945, in such a way to meet the global regulatory requirements and are in accordance with the regulatory requirements of other countries like USFDA and EMA etc. Medical Device Rules, 2017 is not limited to the manufacturer but also provide requirements for import of small quantity of medical devices for personnel use and for the purpose of clinical investigational purpose by the institute deals with the research and development of medical devices. This document provides guidance to assist manufacturers, traders/distributors, clinical establishments, healthcare professionals and general public on nationally recognized regulatory requirements concerning medical device in India.
Introduction
Medical devices are an important part of health care, yet they are an extraordinarily heterogeneous class
of products. The term medical device includes such technologically simple items as ice bags and tongue
depressors on one end of the continuum and very sophisticated items such as cardiac pacemakers and proton
therapy devices on the other end. Broadly based on the function of medical device they may be classified as
preventive care device, assistive care device, diagnostic device and therapeutic device. Perhaps these are the
unique challenges like safety concerns and diversity of products coupled with the sheer number of different
devices in market that makes the development of an effective and efficient regulatory scheme a unique
challenge for domestic as well as international regulatory bodies. Regulators and governments count on
standards to help develop better regulation.
CDSCO has made tremendous progress in health sector, framing laws and setting standards for medical devices remained a big challenge. This assumes importance especially when the medical devices impact health of the patients. Therefore, the Govt. of India, Ministry of Health & Family Welfare has recently notified the Medical Devices Rules, 2017 [1] and made the same as part of the Drugs and Cosmetics Act, 1940. Medical devices are used as diagnostics and also for treatment of diseases. Therefore, like other pharmaceutical products, the quality of medical devices is also required to be monitored for ensuring efficacy and patient safety [2].
Medical devices are being widely used in all branches of medicines, surgery and community not only in India but across the globe. Keeping in view the broad objectives for ensuring protection of the health & safety of patients, healthcare professionals and others, the Ministry of Health & Family Welfare, Government of India has released the Medical Device Rules, 2017, effective from 1st January, 2018 for regulating Medical Devices being used in the country. As India is playing a major role in marketing of these devices in Asia, and beyond, regulating Medical Devices poses a real challenge, upon implementation of the Medical Device Rules, 2017 which ultimately aim at replacing the existing Rules of the Drugs and Cosmetics Act, 1940 [3].
Classification of Medical Devices [4]
Medical devices are classified in different classes which includes
1. A non-invasive medical device which comes into contact with injured skin. It is intended to be used as a
mechanical barrier, for compression / for absorption of exudates only.
2. Intended for channeling or storing body liquids / tissues / liquids / gases for the purpose of final infusion,
administration / introduction into a human body.
1. Comes in contact with injured skin to intend management of microenvironment of wound.
2. If it is intended to be connected to active medical device, which is in “Class B, C or D” or for channeling
blood or storing or channeling other body liquids/storing organs, parts of organs / body tissues
3. If the intended modification is being carried out by filtration, centrifuging or any exchange of gas or heat.
1. Intended to use with breached surfaces as a healing is a primary intention.
2. Blood bag that does not incorporate a medicinal product.
3. If it is intended for modifying the biological/ the chemical composition of blood/other body liquids /other
liquids intended for infusion into the body.
1. Intended for transient uses and
2. Not intended to be connected to active medical device(s) or
3. Intended to be connected only to a Class A medical device.
4. Intended for use in an oral cavity as far as the pharynx or in an ear canal up to the ear drum or in a nasal cavity.
5. Not liable to be absorbed in mucous membrane.
6. Reusable surgical instrument.
1. Intended for use on the external -surface of an eyeball.
2. Liable to be absorbed in mucous membrane.
3. Intended for short term use.
4. Not intended to connect an active medical device.
5. Intended to be connected to a Class A medical device(s) only.
6. Intended for use in an oral cavity as far as the pharynx / in ear canal up to the ear drum or in a nasal cavity.
7. If it is intended to be connected to active medical device which is in “Class B, C or D”.
8. Surgically invasive devices
9. Devices that can be placed surgically in tooth for short term as well as long term.
1. If it is intended for long term use and, not intended to be connected to active medical device(s) or it is to
be connected to Class A medical device only.
2. If it is intended for the supply of energy in the form of ionising- radiation.
3. If it is intended to have a biological effect or to be wholly or mainly absorbed by the human body.
4. If it is intended for the administration of any medicinal product by means of any delivery system and such
administration is done in a manner that is potentially hazardous.
5. A short-term use surgically invasive medical device is assigned to as Class C, if it is intended to undergo
chemical changes in the body.
6. If it is intended for the administration of any medicinal product or the supply the energy in form of
ionising -radiation.
7. Long term implantable devices.
1. If it is intended to be used specifically in direct contact with the “central nervous system” or for the
diagnosis, monitoring or correction of a defect of the heart or of the “central circulatory system” through
direct contact with these body parts.
2. If it is intended to be have a biological effect or wholly or mainly absorbed by the human body or to be
used specifically in direct contact with the central nervous system (CNS) or for the diagnosis, monitoring
or correction of a defect of the heart or of the central circulatory system (CCS) through direct contact with
these parts of the body.
3. To be used in direct contact with the heart, the central circulatory system or the central nervous system
(CNS), life supporting systems, an active medical device, to be wholly absorbed by human body, for
administration of medicinal product, to be breast implanted and an intended for long term chemical change
in body.
1. If administration or exchange of energy to or with the human body by such medical devices is intended
to be used.
1. If the administration or exchange of energy is to be done in a potentially hazardous way (like, through the
emission of ionising- radiation), taking into account the nature, density and site of application of the energy
and type of technology involved.
2. If it is intended for the control or monitoring, or to be used to directly influence the performance of “Class
C” active therapeutic device.
1. If it is to be used solely to illuminate the body of patient with light in the visible or near infrared spectrum.
1. To supply energy, which will be absorbed by the human body
2. To capture images of the in vivo distribution of radio-pharmaceuticals
3. For the direct diagnosis or monitoring of vital physiological process(s).
4. If it is intended for the administration, or removal of any medicinal -product, body liquid or other substance
to or from a human -body.
1. For the monitor of vital physiological parameters, and nature of any variation is such that it may result in
immediate danger to the patient (such as any variation in cardiac performance, respiration or activity of the
CNS)
2. For the diagnosing in a clinical situation when the patient is at position of immediate danger.
3. If it is intended for the emission of ionising-radiation and to be used in diagnostic or interventional
radiological procedure.
4. If it is intended for the control or monitor, or to directly influence the performance of any active diagnostic
medical device.
5. If the administration or removal of the medicinal product, body liquid or other substance is done in
a manner that is potentially hazardous, taking into account of the nature of the medicinal product/body
liquid/ substance; the part of body concern; the mode and route of administration/removal.
1. These medical devices are assigned to class D in case it incorporates as an integral part a substance which
is considered to be a medicinal-product, act on a human body with action ancillary to that of the medical
device.
2. The device manufactured from or incorporates of cells, tissues or derivatives of cells or tissues, or any
combination thereof, of animal or human origin, which are or have been rendered non-viable or cells, tissues
or its derivatives of cells or tissues, or any combination thereof, microbial /recombinant origin.
1. These are included in class B if it is intended for the disinfection of any other medical device before the
latter is sterilized / undergoes end-point disinfection.
2. If it is intended to be used specifically for the sterilization of any other medical device, the end-point
disinfection of any other medical device and disinfection, cleaning, rinsing or hydration of contact lenses etc.
included in class C
1. A medical device intended to be used for contraception or the prevention of the transmission of any STD
is assigned as “Class C”.
2. If it is an implantable medical device or an invasive medical- device intended for long term use included
in Class D.
Drug-eluting stents are a combination product including both a drug and device used for the treatment of symptomatic coronary artery disease (CAD). The device mimics the scaffolding properties of cardiac stents, but also includes either a coating of drug directly on the stent or a thin polymer coating of anti-proliferative drug intended to inhibit vascular responses to arterial injury and reduce restenosis. DES is a minimally invasive treatment option when percutaneous coronary angioplasty or stenting is required [5]. The products that combines with the medical devices and incorporate elements of a medical device and a drug used for treatment of the any ailment or medical condition of the human or animals and is directly or indirectly related to intended use of the medical device are known as combination products as the drug like sirolums when combined with the cardiac stent by means of formulations and used for treatments of post- surgical conditions, therefore it is categorized as a combination product [6].
Sirolimus, also called Rapamycin, was originally developed in 1975 as a macrolide antibiotic. It has potent antifungal, immunosuppressant, and antitumor properties.
Sirolimus inhibits expression of key cytokines necessary for the smooth muscle cell to progress from the G1 to the S phase of the cell cycle and thus arrests the cell in the G1 phase. Because sirolimus unlike cytotoxic agents, arrests cellular proliferation, it is referred to as a cytostatic agent.
When used as part of a DES system, Sirolimus targets the very cause of ISR, proliferating vascular smooth muscle cells, arresting their proliferation. Since it does not kill the cells, it avoids the inflammation associated with massive cellular necrosis seen with cytotoxic approaches, such as brachytherapy [7].
History of Drug Eluting Medical Devices
Kentucky - No legislative mandates for coverage of drug eluting stents were found.
Indiana - No legislative mandates for coverage of drug eluting stents were found.
Tennessee - No legislative mandates for coverage of drug eluting stents were found.
The FDA granted new device approval to the CYPHER Sirolimus-eluting coronary stent on April 24, 2003.
The FDA is requiring the maker of the CYPHER stent to conduct a 2000-patient post-approval study. The study is to continue evaluation of patients from ongoing clinical trials to assess the long-term safety and effectiveness of the CYPHER stent and to look for rare adverse events that may result from the use of their stent [8].
- The use of balloon angioplasty with or without coronary artery stenting is limited by the phenomenon
of in-stent restenosis (ISR). Until very recently, most efforts to overcome ISR had been ineffective. The
search to prevent or reduce the frequency of ISR has led to the recent development of novel coronary artery
stents designed to deliver a drug that acts locally. The first DES was approved by FDA in April 2003. This
stent releases sirolimus, an agent that inhibits vascular smooth-muscle-cell proliferation. To date, four major
clinical trials have demonstrated the sirolimus eluting stent to be safe and effective in preventing restenosis
in de novo coronary artery lesions [7,9].
The Central Drugs Standard Control Organization (CDSCO) under Directorate General of Health
Services in Ministry of Health and Family Welfare (MoHFW), Government of India (GoI) is the National
Regulatory Authority (NRA) responsible for approval of manufacturing, import, conduct of clinical trials,
laying down standards, sale and distribution of medical devices through enforcement and implementation
of the Medical Devices Rules, 2017 [1] released through Gazette of India notification G.S.R. 78(E), dated
31st January 2017 by the MoHFW, GoI [3].
All the applicants are required to apply to the CDSCO the Central Licensing Authority through online portal at website of cdsco.gov.nic.in, before procedure of the application, applicant is required to be registered to the CDSCO and after completion of the registration process a login ID and password is generated automatically, which is shared through registered email id and mobile number of the firm.
After completion of the registration procedure an applicant can apply through online system of SUGAM portal for Medical Device approval.
Procedure for submission of online application through SUGAM portal
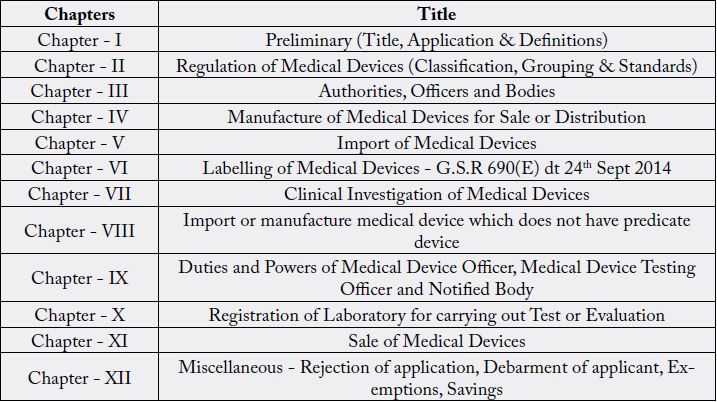
Therefore, all the medical Device manufacturer has to follow the Medical Devices Rules, 2017, which has following chapters and title:
Materiovigilance Programme of India (MvPI)
The NRA is intended to ensure a high level of protection of public health and safety. As NRA, CDSCO
has the responsibility to conduct Materiovigilance programme of India (MvPI). Indian Pharmacopoeia
Commission functions as National Coordination Centre (NCC) for MvPI. Sree Chitra Tirunal Institute
for Medical Sciences & Technology (SCTIMST), Thiruvananthapuram, shall act as National Collaborating
Centre; National Health System Resource Centre (NHSRC), New Delhi, shall act as Technical support
partner, and Central Drugs Standards Control Organisation (CDSCO), New Delhi, shall continue to
function as regulator. Materiovigilance Programme of India (MvPI) is meant to enable safety data collection
in a systematic manner so that regulatory decisions and recommendations on safe use of medical devices
being used in India could be based on data generated here. The programme is meant to monitor medical device-associated adverse events (MDAE), create awareness among healthcare professionals about the
importance of MDAE reporting in India and to monitor the benefit-risk profile of medical devices. It is
also meant to generate independent, evidence-based recommendations on the safety of medical devices and
to communicate the findings to all key stakeholders.
Approval Procedure in India
In India Medical devices and Drugs are regulated under the provisions of the Drugs and Cosmetics Act ,
1940 nd Rules made thereunder.
Medical Devcies are defined under Section 3 (b)(IV) of the Drugs and Cosmetcis Act, 1940
1. Ministry/Department involved:
• Central Drugs Standard Control organization
• Department of Pharmaceuticals
• Ministry of Health & Family Welfare
2. Associations involved:
• ADMI -Association of Diagnostics Manufacturers of India
• AIMED -Association of Indian Medical Device Industry
• MTAI-Medical Technology Association of India
The medical device market is having the steadily growth in India. It was valued around US$3.5 billion in
2015 and may expand to approximately US $4.8 billion in the year 2019, its medical device market emerges
as a promising opportunity for foreign manufacturers, due to progress in India economy, healthcare, and
social landscapes [10].
The Central Drugs Standard Control organization (CDSCO) is an Indian regulatory agency in central
Government under Department of Health and Family Welfare, and is responsible for regulating cosmetics,
medicines and medical devices being manufactured or import in the country along with the State Drugs
Regulatory Authorities.
The CDSCO has legal procedure as per the Drugs and Cosmetics Act 1940, which is designed to ensure that Drugs or medical devices supplied in India or exported to the other countries shall have the acceptable standards of quality, safety and efficacy (performance). To ensure the benefit to consumers balance any risks associated with the use of medicines and medical devices, these regulations are framed by the CDSCO and are based on scientific and clinical expertise. The CDSCO relies on the public, healthcare professional as well with industries to report problems with medicines or medical device. CDSCO investigates the report(s) received by it to determine any necessary regulatory action.
Earlier, till 2017, the medical devices were being regulated in INDIA as per the schedule M-III of the drugs
and cosmetics Rules, 1945 which provides information about the area requirements, equipment and facilities
to be provided for manufacturing of the medical devices.
To improve the safety, quality and performance of the Medical devise to be imported and manufactured in India. Govt. of India published the draft new medical device Rules under sub section 1 of Section 12 and Sub-section 1 of Section 33 of the Drugs and Cosmetics Act, 1940 “23 of 1940” on 17th october, 2016 vide Gazette Notification no: G.S.R. 983(E)(“Medical Device Rules, 2017 | Regulatory Updates in India,”).
After consultation with the advisory committee i.e. Drugs Technical Advisory Board, these rules are finalized and published as Medical device Rules, 2017 vide GSR 78(E) on 31st January, 2017 and are made effective from January, 2018.
The New Medical device rules are framed by the Ministry of Health and Welfare (MoHFW), Govt. of India so par and equivalent to the regulatory requirements of other developed countries like USA and European Union.
This initiative was taken by the Government of India not to stand as a stringent regulatory Authority with the Global regulatory parameters in the field of Medical Devices but also with a vision to provide the safe, qualitative and effective Medical devices to the Indian and Global patients.
• Introduction of the Medical Device classification on risk-based approach and Parameters classification.
• Roles and responsibilities of the Licensing Authority of State and Licensing Authority of Central for
regulation of Medical Device as per the Classification.
• Notification and registration of the Notified Bodies for Inspection of the Medical Devices.
• Provisions of Test License to manufactures/ Importers for test- evaluation and clinical investigations etc.
• Import of medical devices, investigational medical device by Government hospitals or medical institutions
for treatment of patients.
• Imports of medical - device for personal use.
• Unique device identification-UDI of the medical device.
• Clinically investigations of medical device and performance evaluation of new in vitro diagnostic medical
device.
• Compensation and Medical management related to clinical investigation.
• Imports /manufacturing of medical device, which does not have any predicate device.
• Registration of testing -laboratory for carrying out test or evaluation
• Recall of medical- devices.
Classification of Medical-Devices in India
As defined in other developed country by their regulatory authorities like USFDA, MHRA and EMA,
CDSCO also classified medical devices in new medical device regulations, 2017 as follows:
• Low risk (Class A)
• Low moderate risk (Class B)
• Moderate high risk (Class C)
• High risk- Class D. (Drug Eluting stent)
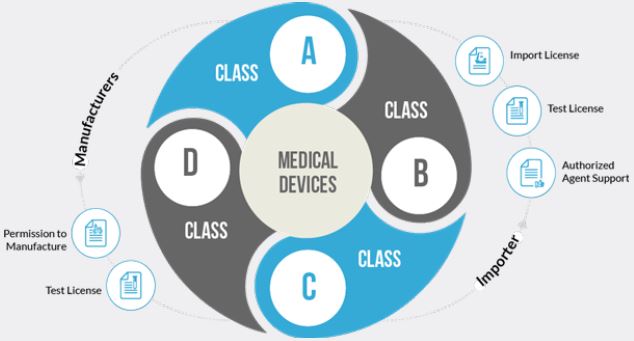
A medical device which itself or its predicate not notified in the medical devices list of CDSCO (Central
Drugs Standard Control organization) is considered a new medical device.
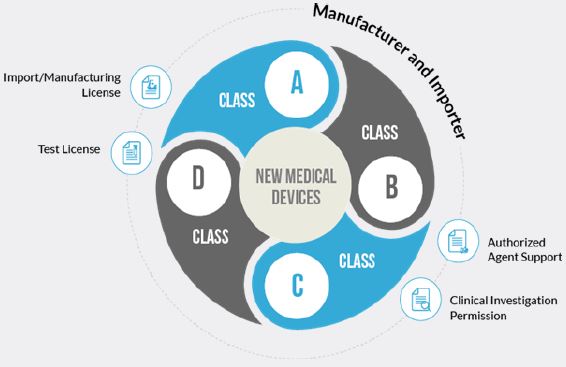
As per Medical Device Rules, 2017, five basic Principles area clarified, and are necessary for the purpose of understanding of the manufacturer before applying to approval of the Medical devise:
The Central Licensing Authority is the Competent Authority for enforcement of these rules in matters relating to- (i) import of all Classes of medical devices; (ii) manufacture of Class C and Class D medical devices; (iii) clinical investigation and approval of investigational medical devices.
Prior submission of the application manufacture has to prepare and define the following parameters with
respect to the drug eluting stent:
All drug eluting stents, mainly which, fall under the category of class C and D and as most of the drug eluting stents are the cardiac stent and are categorized as “Implantable medical devices and surgically invasive medical devices for long term use” are classified as class D medical devices
If the medical device is already approved in the country and having the predicate device, manufacture is required to submit the application in form MD-7 of the medical device(s).
Applicant is required to comply with the requirements of seventh schedule of the medical Device Rules, 2017, in case of investigational medical device(s) and require to apply for permission to manufacture investigational medical device for conduct of clinical investigation in form MD-22 to Central Licensing Authority along with all required data as per seventh schedule (Annexure-6) and require to conduct the clinical evaluation as per requirements of CHAPTER VII i.e.“Clinical investigation of medical device and clinical performance evaluation of new in vitro diagnostic medical device”
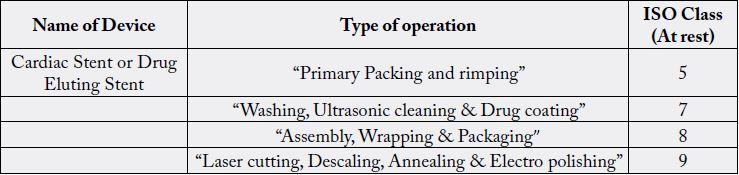
Applicant is required to pay the fee by Challan or by electronic mode, in the Bank of Baroda, Kasturba Gandhi Marg-KGM, New Delhi-11ooo1 or in any other branch of Bank of Baroda, or any other bank which are notified by the Ministry of Health and Family Welfare in the Central Government, to be credited under the Head of Account HoA “o21o- Medical and Public Health, o4-Public Health, 1o4-Fees and Fines along with application and also submit the scanned copy of the receipt of fee at the time of online application in Sugam portal.
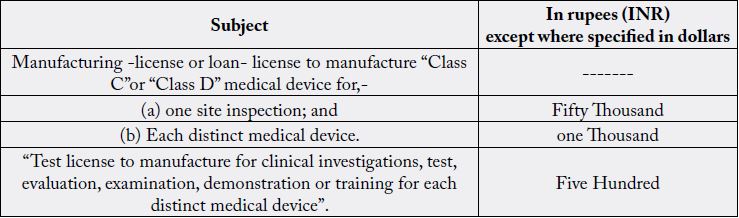
1. Responsibilities of a Medical Device Manufacturer
For each medical device, manufacturer shall determine the following
• Classification of Medical-Devices
• Intended purpose of Medical-Devices
• Appropriate Medical Device Nomenclature- code
• Select and apply appropriate conformity assessment to demonstrate compliance with respect to Essential
Principles
• Ensure for appropriate processes in place and documentation to demonstrate this before submission of
application to the CDSCO for conformity assessment evidence.
• To obtain the conformity assessment evidence and ensure that the information on the certificate remains
current and valid.
• Submission of the application and payment of assessment fees for obtaining the conformity assessment
evidence.
• Preparation of an Indian Declaration of Conformity that includes all the manufacturing details for the
medical devices
• Ensure that their conformity assessment procedures are appropriately maintained once they obtain the
necessary conformity assessment evidence, and that the ongoing requirements are met e.g. reporting adverse
events, regular quality systems audits.
• Notify to the CDSCO regarding substantial changes to the design, production or intended performance
of the device.
Documents to Be Submitted with the Application for Grant of License to Manufacture Drug Eluting Stent, Class D Medical Device
1. An application to the Central Licensing Authority through an identified online portal of the Central
Government for licence or loan licence to manufacture for sale or for distribution, of Class D medical device
in Form MD-7 or Form MD-8 (in case of loan license) through online by using Sugam portal”.
2. The application in Form MD-7 or Form MD-8 are relating to Class D medical device, accompanied with
a fee as specified in the Second Schedule along with documents as specified in clause (ii) of Part II of the
Fourth Schedule.”
3. The Central Licensing Authority may, wherever required, “Class D medical devices, use the services of any
expert in the relevant field for scrutiny of application and other technical documents”.
4. The scrutiny referred of the application and relevant documents is completed by the Central Licensing
Authority in 45 days from the date of online submission of application.
5. In case, where the documents are found to be complete and in order, the Central Licensing Authority
cause an inspection of the manufacturing site by a team of officers accompanied by experts, as may be
considered necessary
6. The Central Licensing Authority may, where required, avail the services of a Notified Body referred to in
sub-rule (4) of rule 13 for inspecting the manufacturing site.
7. In case, where the documents furnished with the application are not found to be complete and in order,
the Central Licensing Authority shall reject the application and inform the applicant of the reasons for such
rejection electronically.
1. Constitution details of domestic manufacturer;
2. Site or plant master file as specified in Appendix I of this Schedule
3. Device master file as specified in Appendix II for medical devices other than in vitro diagnostic medical
devices, or Appendix III for in vitro diagnostic medical devices of this Schedule;
4. Essential Principles checklist for demonstrating conformity to the Essential Principles of Safety and
Performance of the Medical Device including in vitro diagnostic medical device;
5. Test licence obtained for testing and generation of quality control data (for domestic manufacturers only),
if any;
6. Undertaking signed stating that the manufacturing site is in compliance with the provisions of the Fifth
Schedule.
7. Documents as specified in the clause (b) of paragraph (i) of this part.
8. In case of in vitro diagnostic medical devices, a copy of performance evaluation report issued by the central
medical device testing laboratory or medical device testing laboratory registered under sub-rule (3) of rule
83.
While making application for grant of licence or loan licence, the applicant shall meet the following requirements:
1. The manufacturing site shall comply with the requirements of the Quality Management System as
specified under the Fifth Schedule;
2. Appoint competent technical staff under whose direction and supervision the manufacturing activity of a
medical device shall be undertaken and such staff shall possess the following educational qualification and
experience,- (a) degree in engineering in relevant branch or in pharmacy or in science in appropriate subject
from a recognised University and shall have experience of not less than two years in manufacturing or testing
of medical devices; or (b) diploma in engineering (in relevant branch) or in pharmacy from a recognised
institute and shall have the experience of not less than four years in manufacturing or testing of medical
devices;
3. Appoint competent technical staff with degree or diploma in engineering (in relevant branch) or in
pharmacy or in science in relevant subject and having experience of not less than two years in testing of
medical devices under whose direction and supervision, the testing activity of a medical device shall be
undertaken.
Before grant of licence to manufacture for sale or for distribution in respect of Class C or D medical device,
the manufacturing site is inspected within a period of sixty days from the date of application by a team
comprising not less than two Medical Device officers which may include any officer senior to the Medical
Device officer with or without an expert, or a Notified Body.
Provided that no inspection of a medical device manufacturing site for grant of loan licence to manufacture such medical device shall be required to be carried out if the manufacturing site is already Licenced to manufacture such medical device for sale or for distribution.
After completion of inspection, the inspection team forward a descriptive report containing findings on each
aspect of inspection along with the recommendations to the Central Licensing Authority, through online portal of the Ministry of Health and Family Welfare in the Central Government and forward a copy of the
same to the applicant.
(1) Central Licensing Authority, after receipt of the report, and such further enquiry, if any, is satisfied that
the requirements of Rules have been complied, grant a licence in Form MD-9, or loan licence in Form MD-
1o or may reject the application for reasons to be recorded in writing, within a period of forty five days from
the date the inspection report has been received.
(2) In case, a licence or loan licence intends to manufacture additional medical devices in the licensed
manufacturing site, an application for grant of permission to manufacture such medical devices can be
submitted to the Central Licensing Authority along with the fee as specified in the Second Schedule and
the documents as referred to in rule 20 or rule 21.
1. Licence shall be produced, when requested by the Medical Device officer or any other senior officer under
the control of Central Licensing Authority or State Licensing Authority.
2. The licence holder shall inform the State Licensing Authority or the Central Licensing Authority, as the
case may be, of the occurrence of any suspected unexpected serious adverse event and action taken thereon
including any recall within fifteen days of such event coming to the notice of licence holder.
3. The licence holder shall obtain prior approval from the Central Licensing Authority, before any major
change as specified in the Sixth Schedule is carried out and the Central Licensing Authority or the State
Licensing Authority, as the case may be, shall indicate its approval or rejection within forty five days and in
case where no communication is received within the stipulated time from such Authority, such change shall
be deemed to have been approved;
4. The licence holder shall inform any minor change as specified in the Sixth Schedule to the Central
Licensing Authority, within a period of thirty days after such minor change take place;
5. The licence holder shall carry out test of each batch of product manufactured prior to its release for
compliance with specifications either in his own laboratory or in any other laboratory registered under subrule
(3) of rule 83;
6. The licence holder shall, on being informed by the Central Licensing Authority, that any part of any lot
of the medical device has been found not conforming with the provisions specified under the Act and these
rules, and on being directed so to do by such licensing authority, withdraw the remainder of that lot from
sale and, so far as may, in the particular circumstances of the case, be practicable, recall the issues already
made from that lot.
7.The licence holder shall maintain an audit or inspection book in Form MD-11to enable the Notified Body
or Medical Device officer to record his observations and non-conformity, if any;
8. The licence holder shall maintain at least one unit of sample from each batch of invasive medical device
and in vitro diagnostic medical device manufactured for reference purpose for a period of one hundred and
eighty days after the date of expiry of such batch.
9. The licence holder shall maintain records of manufacturing and sales which shall be open to inspection by
a Medical Device officer.
10. The medical device, when offered for sale, shall be accompanied by either its package inserts or user
manual, wherever applicable.
11. The manufacturing or testing activity of medical device shall be undertaken only under the direction and
supervision of the competent technical staff.
12. If the manufacturer has stopped manufacturing activity or closed the manufacturing site for a period of
thirty days or more, the same shall be intimated to the Central Licensing Authority.
In case of change in constitution of a licensee, after grant of licence, the manufacturer inform the Central
Licensing Authority, within forty five days and make an application for grant of licence within a period of
one hundred eighty days from the date of such change in constitution and existing licence is deemed to be
valid till such time, a fresh licence is issued or application is rejected by the Central Licensing Authority.
(1) A licence or loan licence issued in Form MD-9 or Form MD1o is valid in perpetuity, subject to payment
of licence retention fee as specified in the Second Schedule before completion of the period of five years
from the date of its issue, unless, it is suspended or cancelled by State Licensing Authority or the Central
Licensing Authority, as the case may be.
(2) If the licence holder fails to pay the required licence retention fee on or before due date, the licence
holder, in addition to the licence retention fee, be liable to pay a late fee calculated at the rate of 2%. of the
licence retention fee for every month or part thereof within one hundred and eighty days and in the event of
non- payment of such fee during that period, the licence shall be deemed to have been cancelled.
Online Checklist of Documents Required in Sugam Portal Along with Application
1. Covering Letter
2. Constitution of the Firm
3. The Establishment /Site ownership /Tenancy Agreement
4. Copy of Duly notarized valid copies of Quality Certificate in respect manufacturing site(s), if any
5. Copy of Certificate supporting quality management system (ISO: 13485), if any
6. Plant Master file from the Manufacturer as specified in Appendix 1 of Forth Schedule of Medical Devices
Rules (Part-1 to Part-10)
7. Device Master File from the Manufacturer as specified in Appendix II (only for Medical Devices) of
Forth Schedule of Medical Device Rules. Note: In case of Class A devices, Appendix II is not required. For
Class A devices upload information as specified in Part II of Forth Schedule for Medical Devices or IVDs.
8. Performance Evaluation Report of IVDs only
9. Test License obtained for testing and generation of quality control data
10. Undertaking signed stating that the manufacturing site is in compliance with provision of Fifth schedule
11. Fee Challan
12. Legal Form
Post Approval Changes
Post Approval Changes, described in the Sixth Schedule of the Medical Device Rules, 2017 [9] are of two
categories and are required to be intimated to the Licensing Authority.
Changes in respect of following shall be considered as major change in:
1. Material of construction;
2. Design which shall affect quality in respect of its specifications, indication for use; performance and
stability of the medical device;
3. The intended use or indication for use;
4. The method of sterilization;
5. The approved Shelf life;
6. The name or address of,- (i) the domestic manufacturer or its manufacturing site; (ii) overseas manufacturer
or its manufacturing site (for import only); (iii) authorized agent (for import only);
7. Label excluding change in font size, font type, color, labels design;
8. Manufacturing process, equipment or testing which shall affect quality of the device;
9. Primary packaging material.
Changes in respect of following shall be considered as minor change in,
1. Design which shall not affect quality in respect of its specifications, indication for use, performance and
stability of the medical device;
2. In the manufacturing process, equipment, or testing which shall not affect quality of the device.
3. Packaging specifications excluding primary packaging material.
Labelling of Medical Devices
Labeling of medical devices as per Seventh Schedule of Medical Device Rules, 2017 [9].
The following particulars shall be printed in indelible ink on the label, on the shelf pack of the medical device or on the outer cover of the medical device and on every outer covering in which the medical device is packed, namely
1. Name of the medical device;
2. The details necessary for the user to identify the device and its use;
3. The name of manufacturer and address of manufacturing premises where the device has been manufactured;
4. The correct statement about the net quantity in terms of weight, measure, volume, number of units, as the
case may be, and the number of the devices contained in the package expressed in metric system;
5. The month and year of manufacture and expiry (alternately the label shall bear the shelf life of the product):
6. In case of sterile devices, the date of sterilization may be given as date of manufacture of the device.
7. The date of expiry may not be necessary, where the device is made up of stable materials such as stainless
steel or titanium, and supplied non-sterile or in case of medical equipment or instruments or apparatus.
Quality Management System
QMS is applicable to the manufacturer of Finished Medical Device. The manufacturer shall establish,
document, implement and maintain a quality management system and maintain its effectiveness in
accordance with the requirements of QMS.
1. Identify the processes needed for the quality management system and their application throughout the
organization.
2. Determine the sequence and interaction of these processes.
3. Determine criteria and methods needed to ensure that both the operation and control of these processes
are effective.
4. Ensure the availability of resources and information necessary to support the operation and monitoring
of these processes.
5. Monitor measure and analyses these processes.
6. Implement actions necessary to achieve planned results and maintain the effectiveness of these processes.
These processes shall be managed by the manufacturer in accordance with the requirements of this Schedule.
Where a manufacturer chooses to outsource any process that affects product conformity with requirements,
the manufacturer shall ensure control over such processes. Control of such outsourced processes shall be
identified within the quality management system.
1. Documented statements of a quality policy and quality objectives.
2. A quality manual;
3. Documented procedures required by this schedule;
4. Documents needed by the manufacturer to ensure the effective planning, operation and control of its
processes;
5. Records required by this schedule.
6. Where it, specifies that a requirement, procedure, activity or special arrangement be “documented”, it shall,
in addition, be implemented and maintained
7. Data may be recorded by electronic data processing systems or other reliable means, but documents and
record relating to the system in use shall also be available in a hard copy to facilitate checking of the accuracy
of the records. Wherever documentation is handled by electronic data processing methods, authorized
persons shall enter or modify data in the computer. There shall be record of changes and deletions. Access
shall be restricted by “passwords” or other means and the result of entry of critical data shall be independently
checked. Batch records electronically stored shall be protected by a suitable back-up. During the period of
retention, all relevant data shall be readily available.
The manufacturer shall establish and maintain a quality manual that includes,
1. The scope of the quality management system, including details of and justification for any exclusion or
non-application or both;
2. The documented procedures established for the quality management system, or reference to them; and
3. A description of the interaction between the processes of the quality management system. The quality
manual shall outline the structure of the documentation used in the quality management system.
The manufacturer shall prepare documentation in a form of a Plant Master File containing specific information about the facilities, personnel and other details as prescribed in Annexure B appended to this Schedule.
A documented procedure shall be established to define the controls needed.
1. To review and approve documents for adequacy prior to issue;
2. To review and update as necessary and re-approve documents;
3. To ensure that changes and the current revision status of documents are identified;
4. To ensure that relevant versions of applicable documents are available at points of use;
5. To ensure that documents remain legible and readily identifiable;
6. To ensure that documents of external origin are identified and their distribution controlled; and
7. To prevent the unintended use of obsolete documents, and to apply suitable identification to them if they
are retained for any purpose.
The manufacturer shall ensure that changes to documents are reviewed and approved either by the original approving functionary or another designated functionary which has access to pertinent background information upon which to base its decisions. The manufacturer shall define the period for which at least one copy of obsolete controlled documents shall be retained.
Management shall implement the effective quality management system by maintaining the following
1. Communicating to the employees the importance of meeting customer as well as statutory and regulatory
requirements;
2. Establishing the quality policy;
3. Ensuring that quality objectives are established;
4. Conducting management reviews; and
5. Ensuring the availability of resources
Top management of the manufacturer shall ensure that customer requirements are determined and are met.
Top management of the manufacturer shall ensure that the quality policy is appropriate to the purpose,
commitment to comply with requirements, understood within the manufacturer’s organization and reviewed
for continuing suitability.
At planned intervals, quality management system shall be reviewed by top management of the organization
for improvement and the need for changes to the quality management system.
Manufacturer shall establish
1. Design and development planning.
2. Design and development inputs including output(s) of risk management.
3. Design and development outputs with specific characteristics of the product that are essential for its safe
and proper use.
4. Design and development review to identify any problems and propose necessary actions.
5. Design and development verification to ensure that the design and development outputs have met the
design and development input requirements.
6. Design and development validation, as part of design and development validation, the manufacturer shall
perform clinical evaluations.
7. Control of design and development changes, Design and development changes shall be identified and
records maintained for further review, validation and approval of changes
1. Criteria for selection, evaluation and re-evaluation shall be established in purchasing process.
2.Purchase requirements prior to their communication to the supplier shall be specified in the Purchase
information procedure.
1. The manufacturer shall plan and carry out production and service provision under controlled conditions.
2. Manufacturer shall have an effective and validated procedure Cleanliness of product and contamination
control in the area required for production of Medical device as per Annexure-I.
3. Records of installation and verification performed by the manufacturer or its authorized agent shall be
maintained.
4. In case of sterile medical devices, the manufacturer shall maintain records of the process parameters for
the sterilization process which was used for each sterilization batch. Sterilization records shall be traceable
to each production batch of medical devices.
5. Validation of processes for production and service provision shall also be established, maintained and
reviewed.
6. Control of monitoring and measuring devices
In defining the records required for traceability, the manufacturer shall include records of all components, materials and work environment conditions, if these could cause the medical device not to satisfy its specified requirements. The manufacturer shall require that its agents or distributors maintain records of the distribution of active implantable medical devices and implantable medical devices to allow traceability and that such record are available for inspection. Records of the name and address of the shipping package consignee shall be maintained.
1. There shall be an established internal audit procedure to perform the assessment of the activities carried
out and its comparison with the current guidelines as applicable.
2. Frequency of the internal audit shall be defined
Summary
Medical Device Regulations, 2017 provides detailed information about procedure for filing of application
of Medical Devices to the CDSCO for its approval in India with respect to each category of medical
devices which are defined on the basis of the risk involved and different parameters like intended use,
type of use etc. these regulations also provide the elaborately defined the requirements of facilities, Quality
management system to be followed, regulatory timelines, regulatory requirements and approval procedure,
area requirements with respect to environment condition, Clinical investigation requirements for New
Medical Device/investigational Medical Device, labeling for manufacturing of the Medical device in India.
These Regulations are framed by the Ministry of Health And Family Welfare under the Drugs and Cosmetics Act, 1940 and Rules, 1945, in such a way to meet the global regulatory requirements and are so par with the regulatory requirements of other countries like USFDA and EMA etc.
Medical Device Rules, 2017 [9] is not limited to the manufacturer but also provide requirements for import of small quantity of medical devices for personnel use and for the purpose of clinical investigational purpose by the institute deals with the research and development of medical devices.
Conclusion
The study concludes that, This Document Provides guidance to assist manufacturers, traders/distributors,
clinical establishments, healthcare professionals and general public on nationally recognized regulatory
requirements concerning medical device in India.
This Document also aims to be informative in nature on Drug Eluting Stent- Medical Device as and combination product, its regulatory requirements and approval procedure in India whether on human beings and animals.
Bibliography


Hi!
We're here to answer your questions!
Send us a message via Whatsapp, and we'll reply the moment we're available!