Biography
Interests
Mohamed Magdy1, Ola Gaber2, Ihab Eldessouki2, Mohamed Rahouma3*, Mohamed Kamel3, Abdelrahman Mohamed3 & John Morris2
1Pediatric Oncology Department, 57357 Hospital, Cairo, Egypt
2Vontz Center for Molecular Studies, Hematology-Oncology Department, University of Cincinnati, Ohio, USA
3Surgical Oncology Department, National Cancer Institute, Cairo University, Egypt
*Correspondence to: Dr. Mohamed Rahouma, Surgical Oncology Department, National Cancer Institute, Cairo University, Egypt (email: mhmdrahouma@gmail.com)
Copyright © 2018 Mohamed Rahouma, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
TP53 mutations are among the most common genetic alterations observed in human cancers and its product p53 has a major role in protection of the genome. In acute lymphoblastic leukemia (ALL), around 16.2% had normal karyotype by chromosome banding techniques, while 93% showed a TP53 mutation. Chromosome 17 showed monosomy in the majority of them. In chronic lymphoblastic leukemia (CLL), with median follow up of 8.1 years, 14.8% had TP53 mutations. Subclonal TP53 mutations were mostly missense substitutions in the DNA binding domain (78%). In acute myeloid leukemia (AML), TP53 mutations were frequently detected in patients with therapy related AML with its adverse effect on overall survival in those with complex karyotype. In pediatric ALL [1], TP53 mutations occurred in 11.3%. In B cell precursor ALL cases, 12.4% of cases showed TP53 mutation, commonly missense mutations. TP53 mutations were significant predictors of event free survival and time to relapse (hazard ratio (HR), 2.28; 95%CI, 1.41-3.69).
The present study was to evaluate the chewers has the morphological and pathological changes
among the chewers.
Introduction
The P53 is a tumour suppressor protein and is called guardian of the genome [2]. TP53 mutations are among
the most common genetic alterations observed in human cancers and occur in about 50% of unselected
sporadic tumours [3,4]. P53 protein which is encoded by TP53 located on the short arm of chromosome
(17p13) [5,6] was first discovered in the 1979 and it started to gain importance by the end of 1980s when
TP53 mutation was found to be present in many human cancers [7,8].
Human p53 protein consists of 393 amino acids that form several main domains [9,10]. The N-terminal transcriptional domain, the central DNA binding domain, the tetramerization domain, the regulatory domain and the proline rich region. While the transcriptional domain activates pro-apoptotic genes, the tetramerization domain helps p53 form dimers which then form tetramers which is an essential step in positive regulation of gene expression and the central domain facilitates binding of p53 to the response elements in DNA [11].
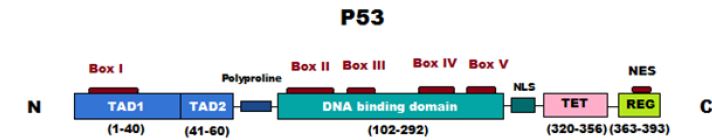
The p53 protein prevents inappropriate cells from proliferation by preventing DNA replication in any condition that may affect the genetic material solidity by inducing apoptosis [12]. Besides p53 role in tumor suppression, it has other roles including DNA damage response, aging, metabolism and fertility [13].
Most of p53 mutations occur in the central domain besides other hot spots in the conserved regions in the gene. When TP53 is mutated in tumor cells, these cells accumulate large amounts of mutant p53 protein. This mutant p53 gains new functions that enhance tumorigenesis, tumor progression and resistance to chemotherapeutic agents and these functions are accomplished by inducing many genes [14-16]. These genes include c-Myc [17], Fos [18], EGF [19], and MDR1 [20].
Acute Lymphoblastic Leukemia (ALL) [21]
Muhlbacher et al. included 808 newly diagnosed ALL patients between 2005 and 2013. Bone marrow or
peripheral blood samples were sent for cytogenetic analysis to the Munich Leukemia Laboratory. They
selected 24 cases with a hypodiploid clone with 40 chromosomes or high hyperdiploid clone with 56-78
chromosomes if the pattern of the lost and retained chromosomes shows that the hyperdiploidy occurred due
to doubling of a hypodiploid clone. 131/808 (16.2%) of the cases showed normal karyotype by chromosome
banding techniques. In 78 cases chromosome banding failed and in 62 of these cases additional comparative
genomic hybridization(CGH) was performed. 5 cases with normal karyotype revealed chromosomal losses
by CGH which was classified to be hypodiploid ALL. FISH analyses confirmed the results of the CGH.
Accordingly, the basis of this study was 29 cases with available bone marrow samples in 25 cases and peripheral blood samples in 12 cases. In the mutational analysis of these patients, 27/29 patients (93%) showed a TP53 mutation. Chromosome 17 showed monosomy in 26/27 of these cases.
There were follow up samples available for 4/29 of those patients. Two patients achieved complete molecular remission during follow up and TP53 was not detectable showing that TP53 mutation was somatic. The third patient did not achieve molecular remission but there was a significant decrease of the TP53 mutation load in follow up samples as it decreased from 76% to 4.76% which was detected by next generation sequencing. The 4th patient did not achieve remission so they could not determine whether TP53 mutation was somatic or germline [21].
Chronic Lymphoblastic Leukemia (CLL) [22]
Rossi et al. published a study in 2014 which was based on 309 newly diagnosed CLL patients who were
prospectively registered in the Amedeo Avogadro University CLL database from 1996 to 2011. The median
follow up of living patients was 8.1 years with no patients lost to follow up.
In this study the aim was to assess the difference in overall survival between cases with wild type TP53 gene and those with small TP53 mutated subclones keeping in mind that the exact prevalence of small TP53 mutated subclones in CLL was unknown.
Screening for TP53 mutation was performed on peripheral blood mono-nuclear cell samples which were collected at diagnosis. Also clonal evolution analysis was performed on peripheral blood samples collected at progression requiring treatment, relapse and last follow up.
Ultra-deep new generation sequencing of TP53 mutation hotspots was performed using the 454 chemistry and based on amplicon libraries. The small TP53 mutated subclones were validated by double step approach, the first step [23] was independent PCR amplification and ultra-deep new generation sequencing experiments while the second step was allele specific PCR [24].
Using ultra-deep new generation sequencing, they could identify 85 TP53 mutations in 14.8% (46/309) of CLL patients and additional 50 subclonal TP53 mutations. Subclonal TP53 mutations were the only TP53 variant in 5.8% (18/309) patients and coexisted in the same population along with TP53 mutation in 3.2% (10/309) of patients. They also considered 17p13 deletion among the genetic defects that target TP53 gene, and those accounted for 4.8% (15/309) patients. So the subclonal mutations were detected solely in 30% (15/50) of all cases carrying TP53 defects in this study.

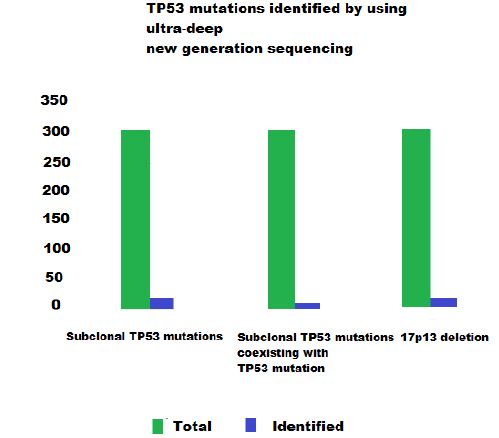
They found that subclonal TP53 mutations were mostly missense substitutions in the DNA binding domain of TP53 protein and they represented 78% (39/50) of all cases carrying TP53 defects. They also found that the median residual transactivational activity of subclonal TP53 mutations towards p21 promoter was only 14.5% compared with wild type TP53 [25]. The rest of the subclonal TP53 mutations were splice site variants representing 10%, nonsense variants representing 8% and indel variants representing the remaining 4%.
They concluded that the functional and molecular profiles of TP53 variants and subclonal TP53 variants were not so different so they came to an understanding that subclonal TP53 mutations have negative impact on TP53 function as clonal TP53 variants.
Rossi et al. [22] found that at presentation, cases carrying only subclonal TP53 mutations shared the same clinical and immunogenic picture with cases carrying clonal TP53 variants and they also carry the same poor prognosis. Cases carrying subclonal TP53 mutations had 5year overall survival of 46.3% (P = .0042) which was close to cases carrying clonal TP53 mutations who had 5year overall survival of 34.6% while cases with wild type TP53 had 5year overall survival of 75.1% (P = .6926). They assessed the dynamics of TP mutated subclones in peripheral blood samples collected from patients before initiating treatment and at relapse (n = 13 patients, 61% treated with immune chemotherapy regimens). They found that the TP53 mutated subclones that were detected before initiating treatment became predominant at time of relapse. They concluded that this may have resulted from the removal of wild type TP53 clones by cytotoxic chemotherapeutic agents which allowed the subclonal TP53 mutations to predominate due to their chemoresistence which resulted in the development of chemorefractory clone. On the other hand there were 2 patients who carried solely TP53 subclones at diagnosis but did not require treatment during follow up and in these 2 patients, the TP53 subclonal mutations did not increase in size during the course of the duration which emphasizes their previous conclusion that stated that early detection of small TP mutated subclones can anticipate the genetic composition of the disease at relapse and the development of chemoresistent phenotype [22].
Acute Myeloid Leukemia (AML) [26]
Many studies have shown that TP53 mutations were frequently detected in patients with therapy related
AML [Alternative genetic pathways and cooperating genetic abnormalities in the pathogenesis of
therapy-related myelodysplasia and acute myeloid leukemia] or AML with complex karyotype [27-29]
Studies regarding the prognostic value of TP53 mutations in AML with complex karyotype patients were
controversial. Rucker et al. [29] showed that TP53 mutation was a poor risk factor for overall survival in
AML patients with complex karyotype while these results could not be shown by Bowen et al. [28]
Hou et al. enrolled 500 adult patients who were newly diagnosed to have de novo AML at the National Taiwan University Hospital who had enough cytopreserved cells for analysis from 1995 to 2008. They investigated TP53 mutation in these patients and its interaction with 17 other genetic abnormalities. Status of TP53 mutation in 131 patients was followed during the clinical course to investigate the stability and pathogenic role of TP53 in AML. Patients who had antecedent hematological disease or therapy related AML were excluded from this study.
PCR and direct sequencing were used for mutation analysis of TP53 exons 3-9 and abnormal sequencing results were confirmed by at least 2 repeated analyses and sequential analysis of TP53 mutations during the course of the disease was performed on 420 samples obtained from 131 patients.
Hou et al. could identify 36 different TP53 mutations in 35 patients, of these 28 mutations were found to be missense mutations, 5 were frame shift mutations, 2 were nonsense mutations and 1 was in-frame mutation.
V311 was found in 3 cases, R175H was found in 2 cases, L194R in 2 cases and all other mutations in only one each while 5 cases had double heterozygous mutations. The remaining 30 cases showed only one mutation. They found that TP53 mutated patients were older with median age of 67 years versus 50 years for wild type TP53 patients (P = 0.0003) .TP53 mutated patients also had lower white blood count, blast and platelet counts than TP53 wild type patients (P < 0.0001 , P< 0.0001, P = 0.0267 respectively). FAB M6 patients were found to have the highest incidence of TP53 mutation than other subtypes. On analyzing the data to understand the association of TP53 mutations with cytogenetic and molecular abnormalities , they found that TP53 mutations were more frequent in patients with unfavorable cytogenetics (46.2%) than those with favorable or intermediate risk cytogenetics (1.2%, P< 0.0001). They also found that incidence of TP53 mutations among patients with normal karyotype was 1.8% while among patients with simple chromosomal abnormalities with one or two changes was 0.5% and patients with complex karyotype had an incidence of 58.8% (P = 0.0001). There was no association between TP53 mutations and t(15;17),t(7;11) or 11q23 translocations in any of the patients while one patient with t(8;21) harbored TP53 mutation as well. TP53 was not found also with +8, +11, +13, +21,-5/del(5q) or -7/del(7q). They also found that 37.1% (13/35) of the patients who had TP53 mutations showed additional molecular abnormalities at diagnosis. When comparing incidence of NPM1, FLT3/ITD and DNMT3A between patients with mutant TP53 and wild type TP53, they found that patients with mutant TP53 had lower incidence of NPM1 mutation (2.9% vs 21.9%, P = 0.0041), FLT3/ITD (0% vs 24.3%, P=0.0002) and DNMT3A (2.9% vs 14.8%, P = 0.045) mutations than those with TP53 wild type while there was no difference in incidence of other molecular abnormalities between patients with mutant and wild type TP53.
Sequential studies to follow TP53 mutations were done serially in 420 samples obtained from 131 patients. 126 patients did not have TP53 mutations while only 5 patients had TP53 mutation at diagnosis and had available samples for study. 2 out of those 5 patients who had TP53 mutations could obtain a complete remission(CR). 3 of them lost the original mutation at remission status while 2 patients retained it and those 2 patients relapsed and died of uncontrolled disease. Hou et al. could also detect the original mutation at relapse in the 3 patients who had available samples for serial study at relapse, but they could not detect it in 1 patient.
Pediatric ALL [1]
The fourth most frequent diagnosis in childhood cancer is relapsed ALL affecting about 20% of children
with ALL [30,31]. There was a gradual improvement in the outcome of the relapsed patients by repeated
trials [32-34].
In their study, Krentz et al. aimed to assess the frequency and prognostic value of TP53 mutations and deletions in first relapse of childhood ALL. They enrolled 265 patients from the German multicenter ALLREZBFM2002 relapse trial and analyzed pretreatment bone marrow samples (254/265) or peripheral blood (11/265) samples taken at time of relapse after enrichment of mononuclear cells by Ficoll density gradient centrifugation. The median follow up time of patients without subsequent event was 4 years. They amplified and sequenced hotspot exons 5-8 of TP53 gene by PCR [35]. As for patients who solely carried TP53 deletion without other mutations, they analyzed also exons 2-4 and exons 9-11. They also analyzed cell cycle by DNA flow cytometry in leukemic cells from 223/265 patients.
While analyzing patients’ data, they found 218 patients with B cell precursor ALL and 47 with T cell ALL. Krentz et al. could identify TP53 mutations in 11.3% (30/265) of patients. In B cell precursor ALL cases, 12.4% (27/218) of cases showed TP53 mutation, 37% (10/27) of these cases showed TP53 deletion without mutation, while 30% (8/27) showed TP53 mutation without deletion and 33% (9/27) showed TP53 mutation and deletion.
As for the type of TP53 mutations, Krentz et al. found 16 missense mutations, 2 deletions, 2 insertions and 1 splice site mutation. While trying to figure out whether the TP53 mutations emerged at relapse or was already there at the initial diagnosis, they analyzed samples from 23 relapse patients with TP53 mutations and the results showed that 54% (15/28) of the mutations and deletions emerged at time of relapse.
Krentz et al. found that TP53 mutations were strongly associated with morphologic refractoriness to relapse therapy defined as more than 5% leukemic blasts in bone marrow(BM) after 9 weeks of treatment(P = 0.001). Also the Event free survival probability for patients with TP53 mutations was 0.092±0.076 while for wild type TP53 was 0.486 ± 0.041 (P = 0.001) and the overall survival probability for TP53 mutated cases was 0.346 ± 0.093 while for wild type TP53 cases was 0.546 ± 0.043 (P = 0.019).
They also found that TP53 mutations were significant predictors of event free survival and time to relapse (hazard ratio (HR), 2.28; 95%CI, 1.41-3.69). In the intermediate risk group (S2) with minimal residual disease(MRD) included in the model, the same results were achieved (HR, 2.89; 95% CI, 1.39 -5.98) while high risk group (S3/S4) [36] showed less significance (HR, 1.94; 95% CI, 0.98 - 3.48)
While studying the difference in cell cycle distribution of leukemic blasts with and without TP53 mutations, there was a significant increase of S and G2-M phase cells in patients with TP53 mutations compared to those with wild type (P = 0.013; median 11.2% vs 6.9).
Conclusion
TP53 mutations are found in around 50% of sporadic tumors and carry a poor prognosis. Thus, more studies
need to focus on target therapy against these mutations to improve survival in different tumors.
Bibliography


Hi!
We're here to answer your questions!
Send us a message via Whatsapp, and we'll reply the moment we're available!