CPQ Neurology and Psychology (2019) 1:3Review Article
Electron Microscopy of Mitochondrial Pathology in Central
Nervous Diseases. A Review
Orlando J. Castejon
Biological Research Institute, Faculty of Medicine, Zulia University, Maracaibo, Venezuela
*Correspondence to: Dr. Orlando J. Castejon, Biological Research Institute, Faculty of Medicine, Zulia
University, Maracaibo, Venezuela.
Copyright © 2019 Dr. Orlando J. Castejon. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 28 January 2019
Published: 11 February 2019
Keywords: Mitochondrial Pathology; Congenital Hydrocephalus; Mitochondrial Matrix
Abstract
The present review describes the pathology of mitochondria in numerous central nervous diseases.
Three injured mitochondrial morphological patterns are found in the human edematous cerebral
cortex of patients with complicated brain trauma associated with subdural hematomas or hygroma,
brain tumors, vascular anomalies, and congenital hydrocephalus. Swollen clear (SCM), swollen
dense (SDM), and dark degenerated (DDM) mitochondria are described. SCM were predominantly
found in traumatic brain edema. SDM and DDM were frequently observed in sustained permanent
ischemia induced by brain tumors, vascular anomaly and congenital hydrocephalus. SCM exhibit
low electron dense mitochondrial matrix, enlarged intracristal space and continuity of outer and
inner mitochondrial membranes. SDM show high electron dense matrix and swollen intact or
fragmented cristae. DDM display overall high electron density of matrix and mitochondrial
membranes. The role of anoxic-ischemic conditions of brain parenchyma, calcium overload, lipid
peroxidation and reactive oxygen species, glutamate excitotoxicity, cytochrome C release, and nitric
oxide and its derivatives are discussed in relation with mitochondrial dysfunction and nerve cell
death. The injured mitochondrial patterns are considered markers of lethal nerve cell injury.
Introduction
The fine structural changes of mitochondria and the respiratory system in experimental cerebral edema,
and in different neuropathological conditions have been widely reported since the advent of transmission
electron microscopy [1-28].
Jonhson et al. (2000) [29] reported the MELAS syndrome characterized by mitochondrial myopathy,
encephalopathy, and acute focal severe cerebral edema. Hillered and Chang (1989) [30] found brain
mitochondrial swelling induced by arachidonic acid release during cerebral ischemia. Takeuchi et al. (1991)
[31] also demonstrated inhibition of mitochondrial respiration by arachidonic acid during brain ischemia.
Novikov and Naperstnikov (1994) [17] described profound destructive changes of mitochondria in traumatic
brain edema. Numerous studies of neuronal mitochondria after transient and permanent ischemia and anoxia
in experimental animals have reported that mitochondria undergo a sequence of profound alterations in
structure and function, which contribute to cell death [11,15,16,22,32-54]. Mitochondrial dysfunction has
also been implicated in bipolar disorders [55]. Wang et al. (2002) [56] reported mitochondrial swelling after
stretch- induced injury. Castejón and Castejón (2004) [57] described swollen clear, dense and degenerated
mitochondria in cortical biopsies of human edematous cerebral cortex. Dupuis et al. (2004) reported
mitochondrial dysfunction in amyotrophic lateral sclerosis (ALS).
Mitochondrial dysfunction has been widely reported in aging and neurodegenerative diseases, such as
Alzheimer´s, Parkinson’s and Huntington’s diseases (Zhu et al., 2006) [3,47,58-82].
Structural and functional damage of mitochondria is currently found in traumatic brain injuries [83-87],
mitochondrial encephalomyopathies [88], picrotoxin and kainic acid-induced epilepsy and status epilepticus
in rats [89-91], stroke [92], intracerebral hemorrhage [93].
Shukkur et al. (2006) [94] found mitochondrial dysfunction and tau hyperfosforilation in Ts1Cje, a mouse
model form of Down Syndrome.
The goal of the present work is to describe the mitochondrial alterations in the soma, myelinated axons,
dendrites and synaptic endings of neuronal cells, in the soma and processes of glial cells, and at the level
of capillary wall. As a step toward characterizing mitochondria as lethal markers of nerve cell death, this
study has been focused to establish mitochondrial alterations and mitochondrial types during neuronal
injury associated to human complicated brain trauma with extradural or subdural hematomas or hygromas,
brain tumors, vascular anomalies, and congenital hydrocephalus of patients examined in our laboratories of
electron microscopy and clinical neuroscience.
Submicroscopic Morphology of Injured Mitochondria in Brain Trauma, Tumors,
Vascular Anomalies and Congenital Hydrocephalus
Swollen clear and dense mitochondria are generally observed in pyramidal and non-pyramidal neurons, but
swollen clear mitochondria (SCM) predominate in traumatic brain edema, and swollen dense mitochondria
(SDM) are more frequently observed in tumors, vascular anomalies and congenital hydrocephalus (Figs. 1-3).
SCM are characterized by the low electron density of mitochondrial matrix. and are frequently associated with microtubules and smooth, flattened endoplasmic reticulum cisterns.
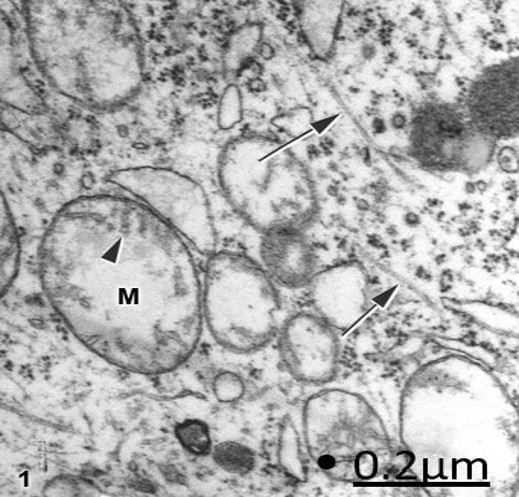 Figure 1: Brain trauma. Subdural hematoma. Right parietal cortex. Ischemic non-pyramidal neuron showing
swollen mitochondria (M) with a clear matrix, fragmented cristae (arrowhead) and intact inner and outer
mitochondrial membranes. The arrows indicate the microtubules. X 60.000.
Figure 1: Brain trauma. Subdural hematoma. Right parietal cortex. Ischemic non-pyramidal neuron showing
swollen mitochondria (M) with a clear matrix, fragmented cristae (arrowhead) and intact inner and outer
mitochondrial membranes. The arrows indicate the microtubules. X 60.000.
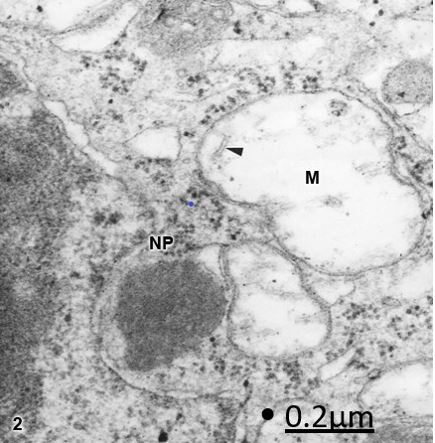 Figure 2: Brain trauma. Subdural hygroma. Left parietal cortex. Non-pyramidal neuron (NP) showing
irregularly shaped and notably swollen mitochondria (M) with clear and swollen matrix, and vestiges of
fragmented cristae (arrowhead). X 90.000.
Figure 2: Brain trauma. Subdural hygroma. Left parietal cortex. Non-pyramidal neuron (NP) showing
irregularly shaped and notably swollen mitochondria (M) with clear and swollen matrix, and vestiges of
fragmented cristae (arrowhead). X 90.000.
Swollen clear and dense mitochondria have been reported in axonal degeneration [95,96], edematous nerve
cells [97], endothelial cells [98], oligodendroglial cells [99], swollen pericites [100,101], reactive astrocytes
[102,103], and degenerated pre- and postsynaptic endings [96,104] in traumatic human brain edema.
According to our studies we have previously postulated the mitochondria as marker of lethal injury [105].
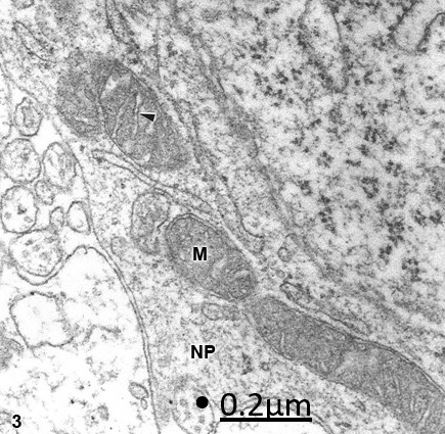 Figure 3: Congenital hydrocephalus. Arnold-Chiari malformation. Right frontal cortex. Non-pyramidal neuron
(NP) showing swollen mitochondria (M) with a dark matrix and swollen, clear, and transversally oriented cristae
(arrowhead). X 60.000.
Figure 3: Congenital hydrocephalus. Arnold-Chiari malformation. Right frontal cortex. Non-pyramidal neuron
(NP) showing swollen mitochondria (M) with a dark matrix and swollen, clear, and transversally oriented cristae
(arrowhead). X 60.000.
Dark degenerated mitochondria (DDM) exhibiting dense matrix, damage of outer mitochondria membrane,
and enlargement of the space between the outer and inner mitochondrial membranes (intermembrane space)
are also found (Fig. 4).
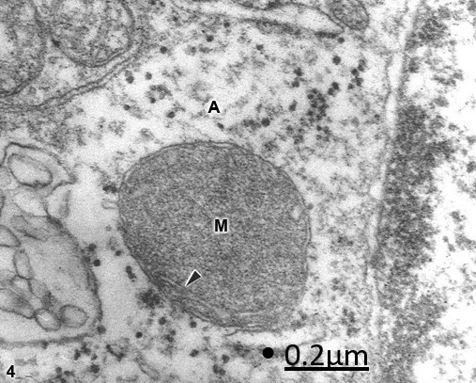 Figure 4: Anomaly of anterior cerebral artery. Right parietal cortex. Swollen astrocyte (A) soma exhibiting a
dense degenerated mitochondrion (M) with few fragmented cristae (arrowhead). X 60.000.
Figure 4: Anomaly of anterior cerebral artery. Right parietal cortex. Swollen astrocyte (A) soma exhibiting a
dense degenerated mitochondrion (M) with few fragmented cristae (arrowhead). X 60.000.
In the swollen astrocytic cytoplasm, SCM, SDM, and DDM are seen, but DDM are predominantly
observed, suggesting a major vulnerability of astrocytic mitochondria to anoxic-ischemic conditions. Clear
and dense mitochondrial populations exhibiting calcium granules, continuous inner and outer mitochondrial
membranes, intact cristae with dilated intracristal space, and fragmentation or absence of mitochondrial
cristae are found in severe brain edema [57].
Marked mitochondrial matrix swelling was earlier reported by Garcia et al. (1977) [9] after middle cerebral
artery occlusion. Mitochondria with increased density of matrix and paracrystalline inclusions have been
reported by Saraiva et al. (1985) [23] in Alzheimer disease. According to Hillered and Chang (1989) [30]
and Takeuchi et al. (1991) [31], brain mitochondrial swelling is induced by the release of arachidonic acid
during ischemia and inhibition of mitochondrial respiration. Mitochondrial swelling and vacuolization was
reported by Ellis et al. (1995) [7] after stretch- induced injury in brain cultured cells. Increased matrix
density of mitochondria and deposit of electron dense material was reported by Solenski et al. (2002) [25]
after severe ischemic/ reperfusion conditions.
At the level of degenerated myelinated axons, SCM and SDM are also distinguished. As illustrated in figure
5, some mitochondria exhibited a dense band occupying the intermembrane space.
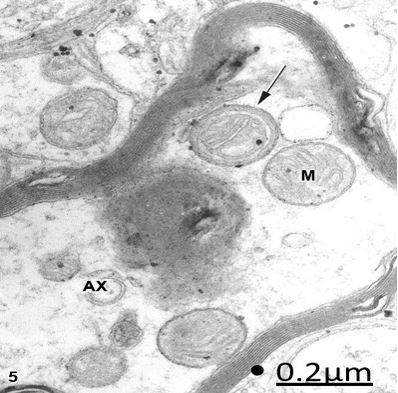 Figure 5: Brain trauma. Subdural hematoma. Left parietal cortex. Degenerated myelinated axon (AX) showing
edematous clear and round mitochondria (M). The arrow points out an unusual mitochondrial morphology
characterized by the presence of a circular dense band beneath the outer mitochondrial membrane. X 60.000.
Figure 5: Brain trauma. Subdural hematoma. Left parietal cortex. Degenerated myelinated axon (AX) showing
edematous clear and round mitochondria (M). The arrow points out an unusual mitochondrial morphology
characterized by the presence of a circular dense band beneath the outer mitochondrial membrane. X 60.000.
In brain trauma, tumors and congenital malformations, varicose swollen dendrites exhibit clear, round and
elongated mitochondria with fragmented cristae (Fig. 6).
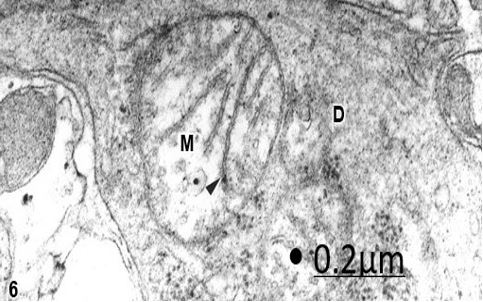 Figure 6: Brain trauma. Contusion and fracture frontal region. Left frontal cortex. Swollen varicose dendrite (D)
showing a swollen mitochondria (M) with a clear matrix and fragmented cristae (arrowhead). X 60.000.
Figure 6: Brain trauma. Contusion and fracture frontal region. Left frontal cortex. Swollen varicose dendrite (D)
showing a swollen mitochondria (M) with a clear matrix and fragmented cristae (arrowhead). X 60.000.
Mitochondrial abnormalities in cortical dendrites also were reported by Paula-Barboza et al. (1984) in early
forms of subacute sclerosing panencephalitis.
At the level of axosomatic synapses upon non-pyramidal nerve cells, vacuolated SDM in the presynaptic
endings and SCM with discontinuities of mitochondrial membranes in the postsynaptic soma surface are
seen in brain tumors (Fig. 7).
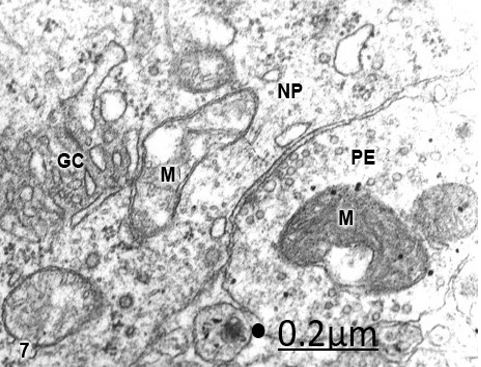 Figure 7: Cystic craniopharyngioma. Right temporal cortex. Axosomatic synapse of a non-pyramidal neuron (NP)
showing dark swollen mitochondrion (M) in the presynaptic ending (PE), and clear swollen mitochondria (M)
with fragmented cristae in the postsynpatic somatic surface. Note the fragmented Golgi complex (GC). X 60.000
Figure 7: Cystic craniopharyngioma. Right temporal cortex. Axosomatic synapse of a non-pyramidal neuron (NP)
showing dark swollen mitochondrion (M) in the presynaptic ending (PE), and clear swollen mitochondria (M)
with fragmented cristae in the postsynpatic somatic surface. Note the fragmented Golgi complex (GC). X 60.000
At the level of the neuropil, the astrocytic processes show a heterogeneous population of mitochondria with
evident signs of calcium sequestration (Fig. 8).
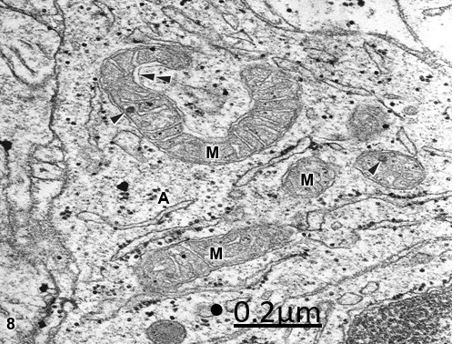 Figure 8: Anomaly of anterior cerebral artery. Right parietal cortex. Interfascicular astrocytic cytoplasm (A)
showing round, elongated, and horse-shoe shaped mitochondria (M). The arrowheads indicate calcium granules.
Note the dense mitochondrial matrix, and the dilated intermembrane space (double arrowhead). X 60.000.
Figure 8: Anomaly of anterior cerebral artery. Right parietal cortex. Interfascicular astrocytic cytoplasm (A)
showing round, elongated, and horse-shoe shaped mitochondria (M). The arrowheads indicate calcium granules.
Note the dense mitochondrial matrix, and the dilated intermembrane space (double arrowhead). X 60.000.
Calcium accumulation in mitochondria was reported by Gutierrez-Diaz et al. (1985) [11] and Hossman et
al. (1985) [12] after cerebral ischemia, and by Hagberg (2004) [43] after hypoxia -ischemia.
The perivascular astrocytic end-feet display also SCM, SDM, and DDM. In severe edema associated to
brain trauma and tumors, isolated astrocytic processes surrounded by proteinaceous edema fluid are observed
showing SCM, SDM, and DDM [57]. Dense degenerated mitochondria are observed in congenital
hydrocephalus at the level of perivascular astrocytic end-feet (Fig. 9).
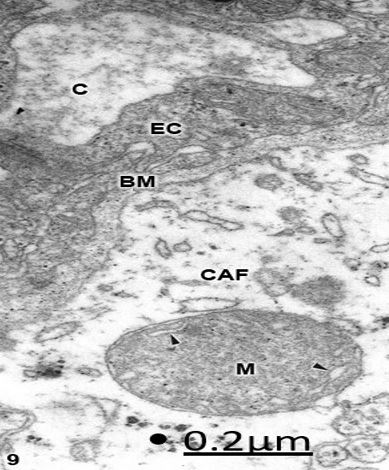 Figure 9: Congenital hydrocephalus and lumbar meningomyelocele. Right parietal cortex. Swollen clear
perivascular astrocytic end-foot (CAF) showing a degenerated mitochondrion (M) with a high electron dense
matrix and few swollen clear cristae (arrowheads). The capillary lumen (C), the endothelial cell (EC) and the
basement membrane (BM) are also distinguished. X 60.000.
Figure 9: Congenital hydrocephalus and lumbar meningomyelocele. Right parietal cortex. Swollen clear
perivascular astrocytic end-foot (CAF) showing a degenerated mitochondrion (M) with a high electron dense
matrix and few swollen clear cristae (arrowheads). The capillary lumen (C), the endothelial cell (EC) and the
basement membrane (BM) are also distinguished. X 60.000.
Swollen clear mitochondria have been reported in congenital hydrocephalus and Arnol-Chiari malformation
[106-108].
In swollen and dark hydropic oligodendroglial cells, SCM and SDM are mainly observed in the soma, and
also in the neighboring astrocytic processes (Fig 10).
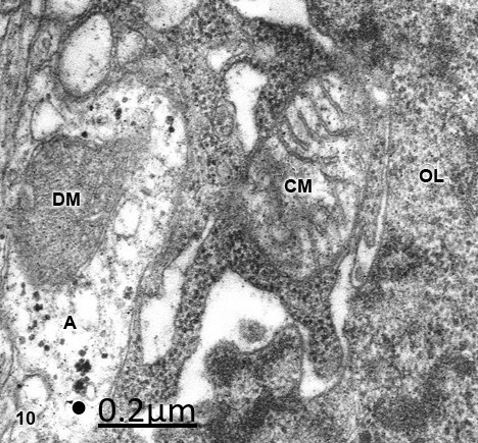 Figure 10: Anomaly of anterior cerebral artery. Right parietal cortex. Oligodendrocyte (OL) showing a swollen
clear mitochondrion (CM) with fragmented cristae and intact outer and inner mitochondrial membranes. On the
contrary, a neighboring clear astrocytic process (A) shows a dense mitochondrion (DM). X 60.000.
Figure 10: Anomaly of anterior cerebral artery. Right parietal cortex. Oligodendrocyte (OL) showing a swollen
clear mitochondrion (CM) with fragmented cristae and intact outer and inner mitochondrial membranes. On the
contrary, a neighboring clear astrocytic process (A) shows a dense mitochondrion (DM). X 60.000.
Al the level of open or collapsed capillaries, SCM and SDM are seen with intact cristae and continuous
inner and outer mitochondrial membrane (Fig 11), mainly localized at the organelle zone of endothelial
cells. DDM are also found in collapsed capillaries with signs of increased transendothelial transport.
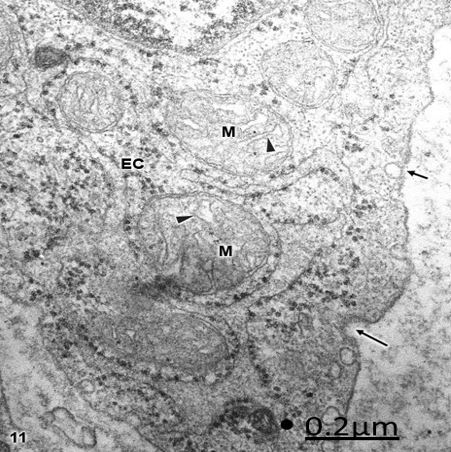 Figure 11: Brain trauma. Subdural hematoma. Right parietal cortex. Endothelial cell (EC) organelle region
showing swollen clear mitochondria (M) with irregularly dilated cristae (arrowheads). Note the formation
of endocytic (short arrows) and clathrin coated (long arrow) vesicles at the level of endothelial cell luminal
membrane. X 60.000.
Figure 11: Brain trauma. Subdural hematoma. Right parietal cortex. Endothelial cell (EC) organelle region
showing swollen clear mitochondria (M) with irregularly dilated cristae (arrowheads). Note the formation
of endocytic (short arrows) and clathrin coated (long arrow) vesicles at the level of endothelial cell luminal
membrane. X 60.000.
Hyperglicemia during cerebral ischemia results in marked changes in endothelial cell morphology and
mitochondrial swelling [109].
The swollen pericytes embedded in the capillary basement membrane exhibit also SCM and SDM (Fig. 12).
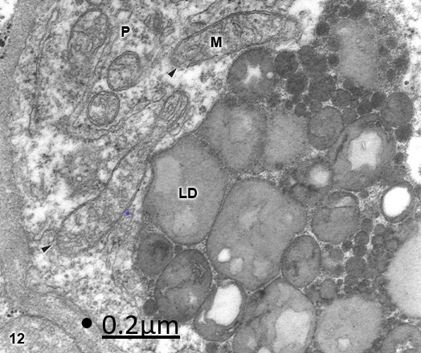 Figure 12: Brain trauma. Subdural hygroma. Right parietal cortex. Pericyte cell (P) showing numerous clear
mitochondria (M) with fragmented cristae and discontinuities (arrowheads) of the outer and inner mitochondrial
membranes. Note the large saturated and insaturated lipid droplet deposit (LD). X 60.000.
Figure 12: Brain trauma. Subdural hygroma. Right parietal cortex. Pericyte cell (P) showing numerous clear
mitochondria (M) with fragmented cristae and discontinuities (arrowheads) of the outer and inner mitochondrial
membranes. Note the large saturated and insaturated lipid droplet deposit (LD). X 60.000.
Swollen clear mitochondria were found in pericyte cells in traumatic brain edema [101].
Light and dense mitochondria have been earlier reported by Ikrenyi et al. (1976) [13] in postmortem
specimens. These authors correlate the light type of mitochondria with the non-functional homogeneous
type, and the dense form of mitochondria with the functional state. More recently, Solenski et al. (2002) [25]
reported dense cortical neuronal mitochondria exposed to severe ischemic/reperfusion conditions, whereas
increasing loss of mitochondrial density with pronounced swelling are observed in permanent ischemia.
Such findings suggest that the SCM type is related with the transient ischemic process of brain trauma,
the formation of subdural hematoma or hygroma, and the associated vasogenic and cytotoxic brain edema.
The SDM type is more frequently found in sustained and permanent nerve cell ischemia, such as in brain
tumors, vascular anomalies and congenital hydrocephalus. In addition, we have found in severe brain edema
enlargement of intracristal space and fragmentation of cristae, which are extension of inner mitochondrial
membrane. The above mentioned findings indicate that mitochondria show significantly different pattern
of injury, express by electron density changes of mitochondrial matrix. This observation is probably related
with mitochondrial matrix protein aggregation, which could be responsible by its osmiophilic property
at the electron microscopy level. The fragmentation of the mitochondrial cristae suggest that oxidative
phosphorylation of ADP, the precursors of the high-energy phosphate bond of ATP, no longer occurs.
In addition, suppose an interruption of mitochondrial membrane intracellular transport, which cause respiration-dependent extrusion of H+ and accumulation of Ca2+ from the cytoplasm (Lehninger, 1971)
[110]. During ischemia the lack of oxygen blocks oxidative metabolism so there is no enough energy to
maintain the membrane potential require to drive Ca2+ uptake into the mitochondria [111]. However,
in brain edema mitochondria can accumulate excessive amounts of Ca2+ and become overloaded. When
this occurs, mitochondria undergo a high permeability transition of the inner mitochondrial membrane
[112,113], release Ca2+, and become swollen and uncoupled [114] thereby loosing the ability to produce ATP,
and leading to nerve cell death. Therefore, neuronal death by necrosis or apoptosis depend of mitochondrial
function [115]. Presumably the different pathological entities examined in the present study induce a high
conductance permeability transition pore or megachannel [116,117] in the inner mitochondrial membrane,
which causes mitochondrial swelling. In this context mitochondrial swelling should be considered as an
epiphenomenon preceding nerve cell death [22].
Glutamate excitotoxicity is the process whereby a massive glutamate release occurs in the central nervous
system in response to ischemia or related trauma leading to a delayed, predominantly ischemic cell death
of neurons. Mitochondria accumulate much of the post-ischemic calcium entering the neurons via the
chronically activated N-methyl-D-aspartate receptors [48,111,118], contributing to excitotoxicity.
Nitric oxide and its derivative peroxinitrite inhibit mitochondrial respiration (complexes I, II and V). NOinduced
inhibition of respiration in brain nerve terminals results in rapid glutamate release, which might also
contribute to neurotoxicity. Peroxinitrite also causes opening of the mitochondrial permeability transition
pore, resulting in release of cytochrome c, which might then trigger apoptosis [119].
Impaired brain mitochondrial respiration and decreased cytochrome oxidase activity have been found after
delayed onset of neurologic deterioration following anoxia/ischemia [27,53]. Novikov and Sharov (1991)
[17] and Sharov and Novikov (1992) [24] reported that the rate of oxidation decrease in mitochondria in
toxic and traumatic edema.
Lipid peroxidation occurs also in brain edema following ischemia and hypoxia [38,120-122], which could
be responsible for the mitochondrial membrane damage. The mitochondrial generation of superoxide anions
is enhanced during anoxia and reoxigenation [118,120]. Mitochondrial electron transport also generates
reactive oxygen intermediates (ROI). A large increase of ROI induce collapse of mitochondrial membrane
potential and neuronal cell death [30,54,120,123]. The elevated intracellular Ca2+ and exposure to fatty
acids, which alter the physical properties of mitochondrial membranes and inhibition of mitochondrial
respiratory components, may enhance this leak of ROI from mitochondria. Cytochrome C is released
from mitochondria into the cytosol contributing to mitochondrial dysfunction and promoting ischemic
neuronal injury and delayed nerve cell death [49,124]. In addition, respiratory inhibition, release of pyridine
nucleotides, and loss of intramitochondrial glutathione necessary for detoxification of peroxides [125].
The amplification of oxidative stress and Ca2+ loading culminates in necrotic cell death [126]. Releasing of
cytochrome C induces to downstream consequences of specific caspase activation and apoptosis [90,127-
129]. Mitochondrial are therefore pivotal regulators of cell death through their role in energy production
and calcium homeostasis, their capacity to release apoptogenic proteins, and to produce reactive oxygen
species [130].
Clinical Neuroscience Studies of Mitochondrial Dysfunction
Mitochondrial dysfunction has been reported in most neurodegenerative diseases. These anomalies include
bioenergetic defect, respiratory chain-induced oxidative stress, defects of mitochondrial dynamics, increase
sensitivity to apoptosis, and accumulation of damaged mitochondria with instable mitochondrial DNA
[131].
Disturbances in mitochondrial functioning are key factors leading to neuroinflammation and
neurodegeneration. Mitochondria have been increasingly linked to the pathogenesis of many neurological
disorders, including multiple sclerosis (MS). [132]. The contemporary data indicate that the axonal
degeneration in multiple sclerosis largely results from the activation of Ca2+-dependent proteases and from
misbalance of ion homeostasis caused by energy deficiency. [133]. There is accumulating evidence highlighting
the central role of mitochondrial dysfunction in axonal degeneration and associated neurodegeneration
[134]. Loss of mitochondrial activity leads to primary mitochondrial diseases and may contribute to
neurodegenerative diseases such as Alzheimer’s and Parkinson’s disease. Mitochondria communicate with
the cell through mitochondrial retrograde signaling pathways. These signaling pathways are triggered by
mitochondrial dysfunction and allow the organelle to control nuclear gene transcription [67]. Convincing
evidence demonstrates oxidative stress as a prominent feature in Alzheimer disease (AD) and Parkinson
Disease (PD) and links oxidative stress to the development of neuronal death and neural dysfunction, which
suggests a key pathogenic role for oxidative stress in both AD and PD [135].
Nissanka and Moraes (2018) analyze the current knowledge in the field of mtDNA and neurodegeneration,
the debate about ROS as a pathological or beneficial contributor to neuronal function, bona fide mtDNA
diseases, and insights from mouse models of mtDNA defects affecting the central nervous system.
There are several diseases that have a mitochondrial origin such as chronic progressive external ophthalmoplegia
(CPEO) and the Kearns- Sayre syndrome (KSS), myoclonic epilepsy with ragged-red fibers (MERRF),
mitochondrial encephalomyopathy, lactic acidosis and strokelike episodes (MELAS), Leber’s hereditary
optic neuropathy (LHON), the syndrome of neurogenic muscle weakness, ataxia and retinitis pigmentosa
(NARP), and Leigh’s syndrome [136].
Mutations of novel genes modifying mainly the balance between mitochondrial fusion and fission have been
shown to lead to overlapping clinical phenotypes ranging from isolated optic atrophy to severe, sometimes
lethal multisystem disorders [137]. Emerging Evidence points to an important role for mitochondrial
dysfunction in the pathogenesis and progression of lysosomal storage diseases (LSD-associated
neurodegeneration). Mitochondrial dysfunction in LSD is characterized by alterations in mitochondrial
mass, morphology and function. Disturbed mitochondrial metabolism in the CNS may lead to excessive
production of mitochondrial reactive oxygen species and dysregulated calcium homeostasis. These metabolic
disturbances ultimately result in mitochondria-induced apoptosis and neuronal degeneration [138,139].
Concluding Remarks
The present review analyzes the presence of three injured mitochondrial morphological patterns in the
human edematous cerebral cortex of patients with complicated brain trauma associated with subdural
hematomas or hygroma, brain tumors, vascular anomalies, and congenital hydrocephalus. Swollen clear
(SCM), swollen dense (SDM), and dark degenerated (DDM) mitochondria are described. SCM were
predominantly found in traumatic brain edema. SDM and DDM were frequently observed in sustained
permanent ischemia induced by brain tumors, vascular anomaly and congenital hydrocephalus. SCM exhibit
low electron dense mitochondrial matrix, enlarged intracristal space and continuity of outer and inner
mitochondrial membranes. SDM show high electron dense matrix and swollen intact or fragmented cristae.
DDM display overall high electron density of matrix and mitochondrial membranes. The role of anoxicischemic
conditions of brain parenchyma, calcium overload, lipid peroxidation and reactive oxygen species,
glutamate excitotoxicity, cytochrome C release, and nitric oxide and its derivatives are discussed in relation
with mitochondrial dysfunction and nerve cell death. The injured mitochondrial patterns are considered
markers of lethal nerve cell injury.
Bibliography
- Arai, C. & Ozawa, K. (1965). Studies on the biochemical aspects of brain injuries and brain edema with special reference to functional changes of mitochondria in the brain. Shinkei Kenkyu No Shimpo., 9, 611-622.
- Brown, A. W. & Brierley, J. B. (1972). The earliest alteration in rat neurons and astrocytes after anoxia-ischemia. Acta Neuropathol., (Berlin)., 23(1), 9-22.
- Chang, D. T., Rintoul, G. L., Pandipati, S. & Reynolds, I. J. (2006). Mutant huntingtin aggregates impair mitochondrial movement and trafficking in cortical neurons. Neurobiol. Dis., 22(2), 388-400.
- Choi, S. I., Ju, W. K., Choi, E. K., Kim, J., Lea, H. Z., Carp, R. I., Wisniwski, H. M. & Kim, Y. S. (1998). Mitochondrial dysfunction induced by oxidative stress in the brains of hamsters infected with the 263 K scrapie agent. Acta Neuropathol., 96(3), 279-286.
- Clendenon, N. R. & Allen, N. (1979). Organelle and membrane defects: Lysosomes., mitochondria., and cell membrane. In A. J. Popp R. S., Bourke L. R., Nelson H. A., Kimelberg, K. (Eds.). Seminars in Neurological Surgery. Raven Press, (pp. 115-129).
- Danks, R. A., Dorevitch, M., Cummins, J. T. & Byrne, E. (1988). Mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS): adolescent onset with severe cerebral edema. Aust. N. Z. J. Med., 18(1), 69-72.
- Ellis, E. F., McKinney, J. S., Willoughby, K. A., Liang, S. & Povlishock, J. T. (1995). A new model for rapid stretch-induced injury of cells in culture: characterization of the model using astrocytes. J. Neurotrauma., 12(3), 325-339.
- Frantseva, M., Perez Velazquez, J. L., Tonkikh, A., Adamchik, Y. & Charlen P. L. (2002). Neurotrauma/neurodegeneration and mitochondrial dysfunction. Prog. Brain Res., 137, 171-176.
- Garcia, J. H., Kalimo, H., Kamijyo, Y. & Trump, B. F. (1977). Cellular events during partial cerebral ischemia. I. Electron Microscopy of feline cerebral cortex after middle-cerebral artery occlusion. Virchows Arch. B. Cell Pathol., 25(3),191-206.
- Gromek, A., Majewska, D., Czernicki, Z., Jurkiewicz, J. & Kunicki, A. (1973). Biochemical properties of mitochondria in conditions of experimental brain edema in cats. Bull. Acad. Pol. Sci. Biol., 21(10), 701-708.
- Gutierrez-Diaz, J. A., Cuevas, P., Reimers, D., Dujovny, M., Diaz, F. G. & Ausman, J. I. (1985). Quantitative electron microscopic study of calcium accumulation in cerebral ischemia mitochondria. Surg. Neurol., 24(1), 67-72.
- Hossmann, K. A., Grosse Ophoff, B., Schmidt-Kastner, R., Oschlies, U. (1985). Mitochondrial calcium sequestration in cortical and hippocampal neurons after prolonged ischemia of the cat brain. Acta Neurophatol. (Berlin)., 68(3), 230-238.
- Ikreny, K., Dora, E., Hajos, F. & Kovach, A. G. (1976). Metabolic and electron microscopic studies post mortem in brain mitochondria. Adv. Exper. Med. Biol., 75, 159-164.
- Koizumi, J. & Shiraishi, H. (1970). Fine structural changes of mitochondria in cerebral edema and dehydration. Arch. Histol. Jap., 32(3), 241-249.
- Mayevsky, A. & Ziv, I. (1991). Oscillations of cortical oxidative metabolism and microcirculation in the ischaemic brain. Neurol. Res., 13(1), 39-47.
- Newcomb, R., Pierce, A. R., Kano, T., Meng, W., Bosque-Hamilton, P., Taylor, L., Curthoys, N. & Lo, E.H. (1998). Characterization of mitochondrial glutaminase and amino acids et prolonged times after experimental focal cerebral ischemia. Brain Res., 813, 103-111.
- Novikov, V. E. & Naperstnikov, V. V. (1994). The effect of fenibut on the ultrastructure of the brain mitochondria in traumatic edema and swelling. Eksp. Klin. Farmakol., 57(2), 13-16.
- Novikov, V. E. & Sharov, A. (1991). The effect of GABA-ergic agents on oxidative phosphorylation in the brain mitochondria in traumatic edema. Farmakol.Toksikol., 54(6), 44-46.
- Oyamagi, H. (1970). The respiratory system in cerebral edema. Kobe J. Med. Sci., 16(1), 27-40.
- Paula-Barbosa, M. M., Tavares, M. A. & Borges, M. M. (1984). Mitochondrial abnormalities in cortical dendrites from patients with early forms of subacute sclerosing panencephalitis (SSPE). Acta Neuropathol., 63(2), 117-122.
- Romansky, K. & Stamenov, B. (1995). Ultrastructural study of cerebral cortex and subcortical white matter following ligation of bridging veins in cats. Zentralblatl Neurochirurg., 56(3), 111-116.
- Rosenthal, M., Mumford, P. L., Sick, T. J. & Perez-Pinzon, M. A. (1997). Mitochondrial hyperoxidaton after cerebral anoxia/ischemia. Epiphenomenon or precursor to residual damage? Adv. Exper. Med. Biol., 428, 189-195.
- Saraiva, A. A., Borges, M. M., Madeira, M. D., Tavares, M. & Paula-Barbosa, M. M. (1985). Mitochondrial abnormalities in cortical dendrites from patients with Alzheimer’s disease. J. Submicrosc. Cytol., 17(3), 459-464.
- Sharov, A. N. & Novikov, V. E. (1992). Status of oxidative phosphorylation in brain mitochondria during its toxic and traumatic edema-swelling. Vopr Med. Khum.., 38(5), 24-26.
- Solenski, N. J., di Pierro, C. G., Trimmer, P. A., Kwan, A. L., Helm, G. A. & Helms G. A. (2002). Ultrastructural changes of neuronal mitochondria after transient and permanent cerebral ischemia. Stroke, 33(3), 816-824.
- Vannucci, R. C. & Bruklacher, R. M. (2006). Cerebral mitocondrial redox states during metabolic stress in the immature rat. Brain Res., 653(1-2), 141-147.
- Wagner, K. R., Kleinholz, M. & Myers R. E., (1990). Delayed decreases in specific brain mitochondrial electron transfer complex activities and cytochrome concentrations following anoxia/ischemia. J. Neurol. Sci., 100(1-2), 142-151.
- Zauner, A., Daugherty, W. P., Bullock, M. R. & Warner, D. S. (2002). Brain oxygenation and energy metabolism: part I-biological function and pathophysiology. Neurosurgery, 51(2), 289-301.
- Johnson, L. J., Hanley, D. F. & Thakor, N. V. (2000). Optical light scatter imaging of cellular and sub-cellular morphology changes in stressed rat hippocampal slices. J. Neurosci. Methods., 98(1), 21-31.
- Hillered, L. & Chang, P. H. (1989). Brain mitochondrial swelling induced by arachidonic acid and other long chain free fatty acids. J. Neurosci. Res., 24(2), 247-250.
- Takeuchi, Y., Morii, H., Tamura, M., Hayaishi, O. & Watanabe, Y. (1991). A possible mechanism of mitochondrial dysfunction during cerebral ischemia: inhibition of mitochondrial respiration activity by arachidonic acid. Arch. Biochem Biophys., 289(1), 33-38.
- Anderson, M. F. & Sims, N. R. (1999). Mitochondrial respiratory function and cell death in focal cerebral ischemia. J. Neurochem., 73(3), 1189-1199.
- Bambrick, L., Kristian, T. & Fiskum, G. (2004). Astrocyte mitochondrial mechanisms of ischemic brain injury and neuroprotection. Neurochem. Res., 29(3), 601-608.
- Blomgren, K. & Hagberg, H. (2006). Free radicals, mitocondria, and hypoxia-ischemia in the developing brain. Free Radic. Biol. Med., 40(3), 388-397.
- Bonanni, L., Chachar, M., Jover-Mengual, T., Li, H., Jones, A., Yokota, H., et al. (2006). Zinc-dependent multi-conductance channel activity in mitochondria isolated from ischemic brain. J. Neurosci., 26(25), 6851-6862.
- Chen, D., Minami, M., Henshall, D. C., Meller, R., Kisby, G. & Simon, R. P. (2003). Upregulation of mitochondrial base-excision repair capability within rat brain after brief ischemia. J. Cerebr. Blood Flow Metab., 23(1), 88-98.
- Chen, H., Hu, C. J., He, Y. Y., Yang, D. I., Xu, J. & Hsu, C. Y. (2001). Reduction and restoration of mitochondrial DNA content after focal cerebral ischemia/reperfusion. Stroke, 32(10), 2382-2387.
- Dzhafarov, A. I., Magomedov, N. M., Babaev, Kh. F. & Akhmedova, G. Sh. (1989). Lipid peroxidation in the synaptosomal and mitochondrial fraction of separate brain structures in hypoxia. Biull. Eksper. Biol. Med., 107(3), 305-307.
- Fattoretti, P., Bertoni-Freddari, C., Caselli, U., Paoloni, R. & Meier-Ruge, W. (1996). Morphologic changes in cerebellar mitochondria during aging. Anal. Quant. Cytol. Histol., 18(3), 205-208.
- Fiskum, G. (2000). Mitochondrial particpation in ischemic and traumatic neural cell death. J. Neurotrauma., 17(10), 843-855.
- Gnaiger, E., Kuznetsov, A. V., Reiger, G., et al. (2000). Mitochondrial defects by intracellular calcium overload versus endothelial cold ischemia/reperfusion injury. Transplant Internat., 13(Suppl.), S555-S557.
- Hayashi, T. & Abe, K. (2004). Ischemic neuronal cell death and organellae damage. Neurol. Res., 26(8), 827-834.
- Hagberg, H. (2004). Mitochondrial impairment in the developing brain after hypoxia-ischemia. J. Bioenerg. Biomembr., 36(4), 369-373.
- Iijima, T. (2006). Mitochondrial membrane potential and ischemic neuronal death. Neurosci. Res., 55(3), 234-243.
- Jonas, E. A., Hickman, J. A., Hardwick, J. M. & Kaczmarek, L. K. (2005). Exposure to hypoxia rapidly induces mitochondrial channel activity within a living synapse. J. Biol. Chem., 280(6), 4491-4497.
- Lou, M., Chen, Y., Ding, M., Eschenfelder, C.C. & Deuchi, G. (2006). Involvement of the mitocondrial ATP-sensitive potassium channel in the neuroprotective effect of hyperbaric oxigenation after cerebral ischemia. Brain Res. Bull., 69(2), 109-116.
- Martin, L. J., Branmbrink, A. M., Price, A. C., Kaiser A., Agnew, D. M., Ichord, R. N., & Traystman, R. J. (2000). Neuronal death in newborn striatum after hypoxia-ischemia is necrosis and evolves with oxidative stress. Neurobiol. Dis., 7(3), 169-191.
- Nicholls, D. G. & Ward, M. W. (2000). Mitochondrial membrane potential and neuronal glutamate excitotoxicity: mortality and millivots. Trends Neurosc., 23(4), 166-174.
- Perez-Pinzon, M. A., Xu, G. P., Born, J., Lorenzo, J., Busto, R., Rosenthal, M. & Sick, T. J. (1999). Cytochrome C is released from mitochondria into the cytosol after cerebral anoxia or ischemia. J. Cereb. Blood Flow Metab., 19(1), 39-43.
- Perier, C., Tieu, K., Guegan, C., Caspersen, C., Jackson-Lewis, V., Carelli, V., et al. (2005). Complex I deficiency primes Bax-dependent neuronal apoptosis through mitochondrial oxidative damage. Proc. Natl. Acad. Sci., 102(52), 19126-19131.
- Plesnila, N. (2004). Role of mitochondrial proteins for neuronal cell death after focal cerebral ischemia. Acta Neurochir., 89, 15-19.
- Takeda, Y., Pérez-Pinzón, M. A., Ginsberg, M. D. & Sick, T. J. (2004). Mitochondrial consume energy and compromise cellular membrane potential by reversing ATP synthetase activity during focal ischemia in rats. J. Cereb. Blood Flow Metab., 24(9), 986-992.
- Wagner, K. R., Kleinholz, M. & Myers, R. E., (1990). Delayed onset of neurologic deterioration following anoxia/ischemia coincides with appearance of impaired brain mitochondrial respiration and decreased cytochrome axidaxe activity. J. Cereb. Blood Flow Metab., 10, 417-423.
- Wullner, U., Seyfried, J., Groscurth, P., Beinroth, S., Winter, S., Glechmann, M., et al. (1999). Gutathione depletion and neuronal cell death: the role of reactive oxigen intermediates and mitochndrial function. Brain Res., 826(1), 53-62.
- Kato, T. & Kato, N. (2000). Mitochondrial dysfunction in bipolar disorder. Bipolar Disord., 2(3 Pt 1), 180-190.
- Wang, K. W., Oi, S. T., Yang, Z. H., Wang, Z. G., Zhu, P. F., Yang, K. J., Liu, C. Y. & Huang, L. J. (2002). Ultrastructural features of cultured rat cortical astrocytes with stretch-induced injury. Di Yi Jun Yi Da Xue Xue Bao., 22(8), 687-689.
- Castejón, O. J. (2004). Structural pattern of injured mitochondria in oedematous human cerebellar cortex. Brain Injury, 18, 1107-1126.
- Abou-Sleiman, P. M., Mugit, M. M. & Wood, N. W. (2006). Expanding insights of mitochondrial dysfunction in Parkinson’s disease. Nat. Rev. Neurosci., 7(3), 207-219.
- 59. Aliev, G., Smith, M. A., de la Torre, J. C. & Perry, G. (2004). Mitochndria as a primary target for vascular hypoperfusion and oxidative stress in Alzheimer’s disease. Mitochondrion, 4(5-6), 649-663.
- Atamna, H. (2004). Heme, iron, and the mitochondrial decay of ageing. Ageing Res. Rev., 3(3), 303-318.
- Baloyannis, S. J., Costa, V. & Michmizos, D. (2004). Mitochondrial alterations in Alzheimer’s disease. Am. J. Alzheimers Dis. Other Demenc., 19(2), 89-93.
- Bonda, D. J., Smith, M. A., Perry, G., Lee, H. G., Wang, X. & Zhu, X. (2011). The mitochondrial dynamics of Alzheimer's disease and Parkinson's disease offer important opportunities for therapeutic intervention. Curr. Pharm. Des., 17(31), 3374-3380.
- Caspersen, C., Wang, N., Yao, J., Sosunov, A., Chen, X., Lustbader, J. W., Xu, H. W., Stern, D., Mckhann, G. & Yan, S. D. (2005). Mitochondrial abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. Faseb J., 19(14), 2040-2041.
- Devi, L., Prabhu, B. M., Galati, D. F., Avadhani, N. G. & Anandatheerthavarada, H. K. (2006). Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J. Neurosci., 26(35), 9057-9068.
- Gao, J., Wang, L., Liu, J., Xie, F., Su, B. & Wang, X. (2017). Abnormalities of Mitochondrial Dynamics in Neurodegenerative Diseases. Antioxidants (Basel)., 6(2), (pp. 1-25).
- Hashimoto, M., Rockenstein, E., Crews, L. & Masliah, E. (2003). Role of protein aggregation in mitochondrial dysfunction and neurodegeneration in Alzheimer’s and Parkinson’s diseases. Neuromol. Med., 4(1-2), 21-36.
- Hunt, R. J. & Bateman, J. M. (2018). Mitochondrial retrograde signaling in the nervous system. FEBS Lett., 592(5), 663-678.
- Kang, D. & Hamasaki, N. (2005). Alterations of mitochondrial DNA in common diseases and disease states: aging, neurodegeneration, heart failure, diabetes, and cancer. Curr. Med. Chem., 12(4), 429-441.
- Kazuno, A. A., Munakata, K., Nagai, T., Shimozono, S., Tanaka, M., Yoneda, M., Kato, N., Miyawaki, A. & Kato, T. (2006). Identification of mitochondrial DNA polymorphisms that alter mitochondrial matrix pH and intracellular calcium dynamics. PloS Genet., 2(8), e128.
- Keeney, P. M., Xie, J., Capaldi, R. A. & Bennett, J. P. Jr. (2006). Parkinson’s disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J. Neurosci. 26(19), 5256-5264.
- Kato, T. & Kato, N. (2000). Mitochondrial dysfunction in bipolar disorder. Bipolar Disord., 2(3 Pt 1), 180-190.
- Milakovic, T. & Johnson, G. V. (2005). Mitochondrial respiration and ATP production are significantly impaired in striatal cells expressing mutant huntingtin. J. Biol. Chem., 280(35), 30773-30782.
- Melov, S. (2004). Modeling mitochondrial function in aging neurons. Trends Neurosc., 27(10), 601-606.
- Mugit, M. M., Gandhi, S., and Wood, N. W. (2006). Mitochondria in Parkinson disease: back in fashion with a little help from genetics. Arch. Neurol., 63(5), 649-654.
- Reddy, P. H. & Beal, M. F. (2005). Are mitochondria critical in the pathogenesis of Alzheimer’s disease? Brain Res. Brain Res. Rev., 49(3), 618-632.
- Rego, A. C. & Oliveira, C. R. (2003). Mitochondrial dysfunction and reactive oxigen species in exitotoxicity and apoptosis: implications for the pathogenesis of neurodegenerative diseases. Neurochem. Res., 28(10), 1563-1574.
- Shipper, H. M. (2004). Brain iron deposition and the free radical-mitochondrial theory of ageing. Ageing Res. Rev., 3(3), 265-301.
- Sullivan, P. G., & Brown, M. R. (2005). Mitochondrial aging and dysfunction in Alzheimer’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry., 29(3), 407-410.
- Su, K. G., Banker, G., Bourdette, D. & Forte, M. (2009). Axonal degeneration in multiple sclerosis: the mitochondrial hypothesis. Curr. Neurol. Neurosci. Rep., 9(5), 411-417.
- Swerdlow, R. H. & Khan, S. M. (2004). A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med. Hypotheses., 63(1), 8-20.
- Takuma, K. (2006). Mitochondrial dysfunction and apoptosis in neurodegenerative diseases. Nippon Yakurigaku Zasshi, 127, 349-354.
- Yaang, Y., Gehrke, S., Imai, Y., Huang, Z., Ouyang, Y., Wang, J. W., et al. (2006). Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc. Natl. Acad. Sci., 103(28), 10793-10798.
- Enriquez, P. & Bullock, R. (2004). Molecular and cellular mechanisms in the pathophysiology of severe head injury. Curr. Pharm. Des., 10(18), 2131-2143.
- 84. Lifshitz, J., Friberg, H., Neumar, R. W., Raghupathi, R., Welsh, F. A., Janmey, P., et al. (2003). Structural and functional damage sustained by mitochondria after traumatic brain injury in the rat: evidence for differentially sensitive populations in the cortex and hippocampus. J. Cereb. Blood Flow Metab., 23(2), 219-231.
- Lifshitz, J., Janmey, P. A. & McIntosh, T. K. (2006). Photon correlation spectroscopy of brain mitochondrial populations: application to traumatic brain injury. Exp. Neurol., 197(2), 318-329.
- Marmarou, A., Signoretti, S., Fatouros, P., Aygok, G. A. & Bullock, R. (2005). Mitochondrial injury measured by proton magnetic resonance spectroscopy in severe head trauma patients. Acta Neurochir., 95, 149-151.
- Xiong, Y., Shie, F. S., Zhang, J., Lee, C. P. & Ho, Y. S. (2005). Prevention of mitochondrial dysfunction in post-traumatic mouse brain by superoxide dismutase. J. Neurochem., 95(3), 732-744.
- Mikol, J. & Polivka, M. (2005). Mitochondrial encephalomyopathies. Ann. Pathol., 25, 282-291.
- Acharya, M. M. & Katyare, S. S. (2005). Structural and functional alterations in mitochondrial membrane in picrotoxin-induced epileptic rat brain. Exp. Neurol., 192(1), 79-88.
- Chuang, Y. C., Chang, A. Y., Lin, J. W., Hsu, S. P. & Chan, S. H. (2004). Mitochondrial dysfunction and ultrastructural damage in the hippocampus during kainic acid-induced status epilepticus in the rat. Epilepsia., 45(10), 1202-1209.
- Sleven, H. J., Gibbs, J. E. & Cock, H. R. (2006). The antioxidant N-acetyl-L-cysteine does not prevent hippocampal glutathione loss or mitochondrial dysfunction associated with status epilepticus. Epilepsy Res., 69(2), 165-169.
- Moro, M. A., Almeida, A., Bolanos, J. P. & Lizasoain, I. (2005). Mitochondrial respiratory chain and free radical generation in stroke. Free Radic. Biol. Med., 39(10), 1291-1304.
- Kim-Han, J. S., Kopp, S. J., Dugan, L. L., & Diringer, M. N. (2006). Perihematomal mitochondrial dysfunction after intracerebral hemorrhage. Stroke, 37(10), 2457-2462.
- Shukkur, E. A., Shimohata, A., Akagi, T., Yu, W., Yamaguchi, M., Murayama, M., et al. (2006). Mitochondrial dysfunction and tau hyperphosphorylation in Ts1Cje, a mouse model for Down syndrome. Hum Mol. Gent., 15(18), 2752-2762.
- Castejón, O. J. (1985). Electron microscopic study of central axons degeneration in traumatic human brain edema. J. Submicrosc. Cytol., 17(4), 703-718.
- Castejon, O. J. (2018). Electron microscopic study of synaptic degeneration in human central nervous system. A review paper. J. Adv. Microsc. Res., 13(4), 400-408.
- Castejón, O. J., Valero, C. & Diaz, M. (1997). Light and electron microscope study of nerve cells in traumatic oedematous human cerebral cortex. Brain Injury, 11(5), 363-388.
- Castejón, O. J. (1980). Electron microscopic study of capillary wall in human cerebral edema. J. Neuropathol. Exper. Neurol., 39(3), 296-328.
- Castejón, O. J. & Castejón, H. V. (2000). Oligodendroglial cell behaviour in trumatic oedematous cerebral cortex, a light and electron microscopic study. Brain Injury, 14(4), 303-317.
- Castejón, O. J. (1988). Ultraestructural alterations of human cortical capillary basement membrane in perifocal brain edema. J. Submicrosc. Cytol. Pathol., 20(3), 519-536.
- Castejón, O. J. (2011). Ultrastructural pathology of cortical capillary pericytes in human traumatic brain oedema. Folia Neuropathol., 49(3), 162-173.
- Castejón, O. J. (1999). Ultrastructural pathology of Golgi apparatus of nerve cells in human brain edema associated to brain congenital malformations, tumours and trauma. J. Submicrosc. Cytol. Pathol., 31, 203-213.
- Castejón, O. J. (1999). Astrocyte subtypes in the gray matter of injured human cerebral cortex: a transmission electon microscope study. Brain Injury, 13(4), 291-304.
- Castejón, O. J., Valero, C. & Diaz, M. (1995). Synaptic degenerative changes in human traumatic brain edema. An electron microscopic study of cerebral cortical biopsies. J. Neurosurg. Sci., 39(1), 47-65.
- Castejón O. J. (2008). The thesis of mitochondria as marker of lethal injury in the traumatic human brain edema. An electron microscopic study using cortical biopsies. Acta Microscópica (Venezuela), 17, 16-27.
- Castejón, O. J. (1994). Transmission electron microscope study of human hydrocephalic cerebral cortex. J. Submicrosc. Cytol. Pathol., 26(1), 29-39.
- Castejón, O. J. (2009). Blood-brain barrier ultrastructural alterations in human congenital hydrocephalus and Arnold-Chiari malformation. Folia Neuropathol., 47(1), 11-19.
- Castejón, O. J. (2010). Submicroscopic pathology of human and experimental hydrocephalic cerebral cortex. Folia Neuropathol., 48(3), 159-174.
- Keep, R. F., Andjelkovic, A. V., Stamatovic, S. M., Shakui P. & Ennis, S. R. (2005). Ischemia-induced endothelial cell dysfunction. Acta Neurochir., (Suppl)., 95, 399-402.
- Lehninger, A. L., (1971). Respiration and ATP formation in the mitochondria. In: Bioenergetics. Menlo Park, CA, W. A. Benjamin., Inc., (pp. 73-98).
- Morley, P., Tauskela, J. S. & Hakim, A. M., (1999). Calcium overload. In W. Walz (Ed.). Cerebral Ischemia. Molecular and Cellular Pathophysiology. New Jersey, Humana Press. (pp. 69-104).
- Dubinsky, J. M., Brustovetshky, N., Pinelis, V., Kristal, B. S., Herman, C. & Li, X. (1999). The mitochondrial permeability transition: the brain’s point of view. Biochem. Soc. Symp., 66, 75-84.
- Kristian, T., Watherby, T. M., Bates, T. E. & Fiskum, G. (2002). Heterogeneity of the calcium-induced permeability transition in isolated non-synaptic brain mitochondria. J. Neurochem., 83(6), 1297-1308.
- Maragos, W. F. & Korde, A. S. (2004). Mitochondrial uncoupling as a potential therapeutic target in acute central nervous system injury. J. Neurochem., 91(2), 257-262.
- Ankarcrona, M., Dypkbukt, J. M., Bonfoco, E., Zhivotovsky, B., Orrenius, S., Lipton, S. A., & Nicotera, P. (1995). Glutamate-induced neuronal death: A succession of necrosis or apoptosis depending on mitochondrial function. Neuron, 15(4), 961-973.
- Lemasters, J. J., Nieminen, A. L., Qian, T., Trost, L. C. & Herman, B. (1997). The mitochondrial permeability transition in toxic, hypoxic and reperfusion injury. Mol. Cell Biochem., 174(1-2), 159-165.
- Nakai, A., Shibazaki, Y., Taniuchi, Y., Miyake, H., Oya, A. & Takeshita, T. (2004). Role of mitochondrial permeability transition in fetal brain damage in rats. Pediatric. Neurol., 30(4), 247-253.
- Nicholls, D. G., Budd, S. L., Ward M. W. & Castilho, R. F. (1999). Excitotoxicity and mitochondria. Biochem. Soc. Symp., 66, 55-67.
- Brown, G. C. & Borutaite, V. (1999). Nitric oxide: cytochrome c and mitochondria. Biochem. Soc. Symp., 66, 17-25.
- Du, G., Mouithys-Michkalad, A. & Sluce, F. E. (1998). Generation of superoxide anion by mitochondria and impairment or their functions during anoxia and reoxygenation in vitro. Free Radic. Biol. Med., 25(9), 1066-1074.
- Ginsberg, M. D., Watson, B. D. & Bustos, R. (1988). Peroxidative damage to cell membrane following cerebral ischemia. A cause of ischemic brain injury. Neurochem. Pathol., 9, 171-193.
- Kykens, J. A. (1994). Isolated cerebral and cerebellar mitochondria produce free radicals when exposed to elevated Ca2+ and Na+, Implications for neurodegeneration. J. Neurochem., 63(2), 584-591.
- Smith, W. S. (2004). Pathophysiology of focal cerebral ischemia: a therapeutic perspective. J. Vasc. Interv. Radiol., 15(1 Pt 2), S3-12.
- Kobayashi, T., Kuroda, S., Tada, M., Houkin, K., Iwasaki, Y. & Abe, H. (2003). Calcium-induced mitochondrial swelling and cytochrome c release in the brain: its biochemical characteristics and implication in ischemic neuronal injury. Brain Res., 960(1-2), 62-70.
- Starkov, A. A., Chinopoulos, C. & Fiskum, G. (2004). Mitochondrial calcium and oxidative stress as mediators of ischemic brain injury. Cell Calcium, 36(3-4), 257-264.
- 126. Jacobson, J. & Ducken, M. R. (2002). Mitochondrial oxidative stress and cell death in astrocytes-requirement for stored Ca2+ and sustained opening of the permeability transiton pore. J. Cell Sci., 115(Pt 6), 1175-1188.
- 127. Clendenon, N. R. & Allen, N. (1979). Organelle and membrane defects: Lysosomes., mitochondria., and cell membrane. In A. J. Popp R. S., Bourke L. R., Nelson H. A., Kimelberg, K. (Eds.). Seminars in Neurological Surgery. Raven Press, (pp. 115-129).
- Huang, Z., Hou, Q., Cheung, N. S. & Li, Q. T. (2006). Neuronal cell death caused by inhibition of intracellular cholesterol trafficking is caspase dependent and associated with activation of the mitochondrial apoptosis pathway. J. Neurochem. 97(1), 280-291.
- Zhang, K. M. & Lieao, Z. G. (2004). Mitochondrial apoptotic signaling pathway in neurons following brain injury induced by hypoxia. Fa Yi Xue Za Zhi., 20(3), 178-182.
- Blomgren, K., Zhu, C., Hallin U. & Hagberg, H. (2003). Mitochondria and ischemic reperfusion damage in the adult and in the developing brain. Mitochondria and ischemic reprefusion damage in the adult and in the developing brain. Biochem. Biophys. Res. Commun., 304(3), 551-559.
- Procaccio, V., Bris, C., Chao de la Barca, J. M., Oca, F., Chevrollier, A., Amati-Bonneau, P., Bonneau, D. & Reynier, P. (2014). Perspectives of drug-based neuroprotection targeting mitochondria. Rev. Neurol. (Paris)., 170(5), 390-400.
- Patergnani, S., Fossati, V., Bonora, M., Giorgim, C., Marchi, S., Missiroli S, et al. (2017). Mitochondria in Multiple Sclerosis: Molecular mechanisms of pathogenesis. Int. Rev. Cell Mol. Biol., 328, 49-103.
- Kozin, M. S., Kulakova, O. G. & Favorova, O. O. (2018). Involvement of mitochondria in neurodegeneration in multiple sclerosis. Biochemistry (Mosc)., 83(7), 813-830.
- Van Horssen, J., Witte, M. E. & Ciccarelli, O. (2012). The role of mitochondria in axonal degeneration and tissue repair in MS. Mult. Scler., 18(8), 1058-1067.
- Yan, M. H., Wang, X. & Zhu, X. (2013). Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radic Biol Med., 62, 90-101.
- Valero, T. (2014). Mitochondrial biogenesis: pharmacological approaches. Currm Pharmm Des., 20(35), 5507-5509.
- Sissler, M., González-Serrano, L. E. & Westhof, E. (2017). Recent Advances in Mitochondrial Aminoacyl-tRNA Synthetases and Disease. Trends Mol Med., 23(8), 693-708.
- Saffari, A., Kölker, S., Hoffmann, G. F., Ebrahimi-Fakhari, D. (2017). Linking mitochondrial dysfunction to neurodegeneration in lysosomal storage diseases. J. Inherit. Metab. Dis., 40(5), 631-640.
- Chan, P. H. (2004). Mitochondria and neuronal death/survival signaling pathways in cerebral ischemia. Neurochem. Res., 29(11), 1943-1949.

