Biography
Interests
Nighat Sultana1* & Muhammad Saleem1,2
1Pharmaceutical Research Center, PCSIR Laboratories Complex, Karachi-75280, Pakistan
2Departments of Biotechnology, University of Karachi, Karachi, Pakistan
*Correspondence to: Dr. Nighat Sultana, Pharmaceutical Research Center, PCSIR Laboratories Complex, Karachi-75280, Pakistan.
Copyright © 2019 Dr. Nighat Sultana. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Reinvestigation of the flowers of Alstonia scholaris of Pakistan origin have resulted in the isolation of three triterpenoids, two of the ursane type, 3β-acetate- 24-nor- urs-4, 12-diene ester triterpene (1), 3β -hydroxy- 24-nor-urs-4,12,28-triene triterpene (2) and one of the oleanane type 3,28- β -diacetoxy-5-olea-triterpene (3) together with two known triterpenes α-amyrin acetate (4) and ursolic acid (5). These compounds were analyzed for antioxidant and antiurease inhibition tests. Compounds 2 and 4 were subjected to enzymatic bioassays. Compounds 2 and 4 were found to be antioxidant with IC50 107±0.39 and >500μM respectively, while compounds 2 and 4 were also found to be urease inhibitors with IC50 175 ± 0.26 and 218 ± 0.14μM, respectively.

The genus Alstonia comprises about twelve species. Alstonia scholaris Linn. R. Br. belongs to family Apocynaceae [1], grows throughout India, in deciduous and evergreen forests, also in plains [2,3]. Its timber is a non-durable hardwood, suitable for light indoor construction purposes, pulp and paper production. The wood has been used for school blackboards, hence the name ‘scholaris’. They are milk bearing shrubs or trees, with large, entire, generally whorled leaves, and terminal cymes of white flowers.
The plant Alstonia scholaris has been used in different system of traditional medication for the treatment of diseases and ailments of human beings. It is reported to contain various alkaloids, flavonoids and phenolic acids. The flowers of Alstonia scholaris are known to contain lupeol and β-amyrin [4]. The bark contains alkaloids: ditaine, echitenine, echitamine (ditamine) and echitamidine together with triterpenes β-amyrin and lupeol [5-9]. It has been reported as antimicrobial, antiamoebic, antidiarrhoeal, antiplasmodial, hepatoprotective, immunomodulatory, anti-cancer, antiasthmatic, free radical scavenging, antioxidant, analgesic, anti-inflammatory, anti-ulcer, anti-fertility, anaemia, chronic diarrhea, dysentery, menstrual disorders, malarial fever, colic and acute arthritis and wound healing activities [10-14]. There are also reports available for the traditional use of this plant for its cardiotonic, anti-diabetic and anti-arthritic properties. The bark yields a tonic and antiseptic medicine. A concentrated decoction of trunk bark is used as a wash in furunculosis and impetigo, and as a gargle in dental caries. The bark, leaves and milky exudates of Alstonia scholaris are used in India [1-3].
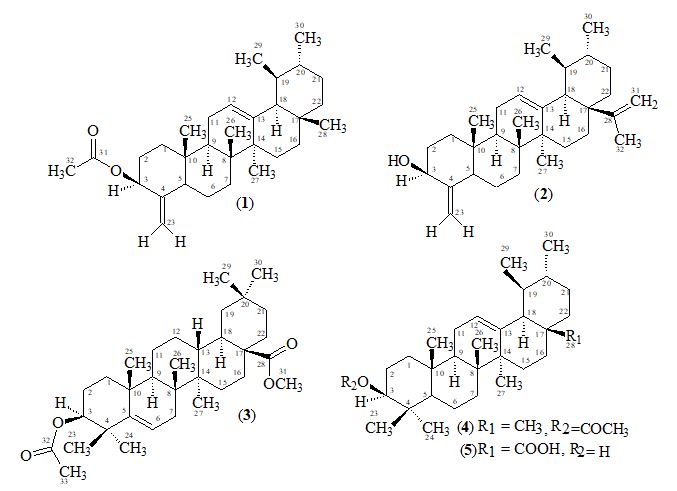
3β -Acetate-24-nor-urs-4,12-diene ester triterpene (1), C31H48O2, was obtained as a white solid mass by column and preparative thin-layer chromatography of the ethanolic extract of A. scholaris flowers.
The molecular composition of 1 was determined as C31H48O2 by high-resolution electron-impact mass measurements of the M+ m/z 452.3652 (calcd. 452.3654) which indicated eight degrees of unsaturation in the molecule.
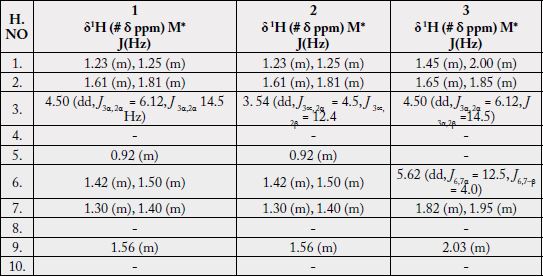
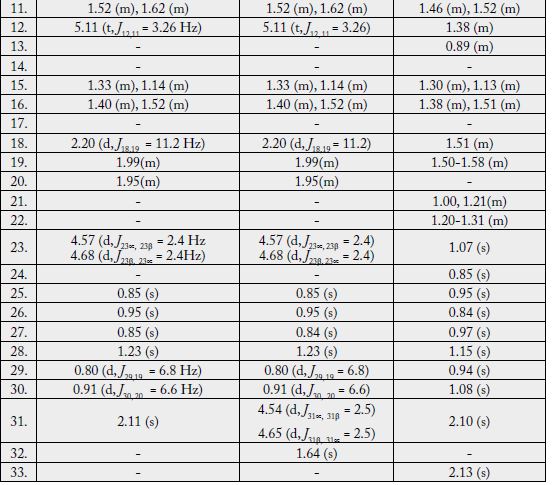
The 1H-NMR spectrum [11,12] (CDCl3, 500MHz) of 1 showed five three-proton singlets (δ 2.11, 0.85, 0.95, 0.84, 1.23) and two methyls as doublets (δ 0.80, J29,19 = 6.5Hz, 0.91, J30,20= 6.5Hz) indicating the presence of five tertiary methyls and two secondary methyls in the molecule as expected in a pentacyclic triterpenoidal skeleton (see experimental section).
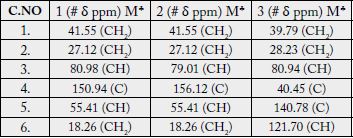
The 13C-NMR spectra (broad-band decoupled and DEPT [11,12] CDCl3, 125MHz) of 1 exhibited signals for all 31 carbons and further supported the formula derived by mass spectrometric observations. DEPT spectra showed the presence of seven methyl, ten methylene, seven methine and (by difference from the broad-band decoupled spectrum) seven quaternary carbons (see experimental section).
Two-dimensional NMR techniques such as COSY 45º, HOHAHA, HMQC and HMBC [12-14] were used to obtain more structural information. The Homonuclear Hartmann Hahn spectrum (HOHAHA) [12-15] recorded with mixing delay of 100ms showed that the C-3 methine proton is coupled with four protons i.e. with the C-2 and C-1 methylenic protons (δ 1.23/1.25 and 1.61/1.81) respectively [16-18]. These observations led to define the structure of 1 as 3β -acetate-24-nor-urs-4,12diene ester triterpene.

3β-hydroxy-24-nor-urs-4,12,28-triene triterpene (2), C31H48O, was obtained as a white amorphous substance by column and preparative thin-layer chromatography of the ethanolic extracts of A. scholaris. The molecular composition was determined as C31H48O by high-resolution electron-impact mass measurements of the M+ (m/z 436). The UV spectrum recorded in methanol showed terminal absorption only, while the infrared spectrum displayed absorptions for hydroxyl (OH, 3415 cm-1) and C = C (1630 cm-1) groups.
The 1H-NMR spectrum [11,12] (CDCl3, 500MHz) of 2 showed four three-proton singlets at δ 0.84, 0.95, 0.85, 1.23 and two methyls as doublets (δ 0.80, d, J = 6.8 Hz, 0.91, J = 6.6 Hz) indicating the presence of four tertiary methyls and two secondary methyls in the molecule as expected in a pentacyclic triterpenoidal skeleton. The downfield region of the spectrum contained only three signals i.e. a broad double doublet at δ 3.54 (J3α,2β = 12.4Hz, J3α,2α = 4.5Hz), four close doublets two at δ 4.57 (d, J23α,23β = 2.4Hz), 4.68 (d, J23β,23α = 2.4Hz) and another two at 4.54 (d, J31α,31β = 2.5Hz) δ 4.65 (d, J31β,31α = 2.5Hz) and a tripplet at δ 5.11 (t, J = 3.26Hz), which could be assigned to the hydroxy-bearing C-3 methine, C-23, 31 methylene and the vinylic C-12 protons, respectively. The chemical shift and coupling constants of the C-3 methine signal indicated an equatorial (β) orientation of the OH group [19].
The 13C-NMR spectra (broad-band decoupled and DEPT [11,12] CD3OD, 100MHz) exhibited signals for all 31 carbons and further supported the formula derived by mass spectrometric observations. The 13C-NMR broad-band spectrum (CDCl3, 100MHz) of 2 showed the resonances of 31 carbon atoms in the molecule. DEPT spectra showed the presence of six methyl, eleven methylene, seven methine and (by difference from the broad-band decoupled spectrum) seven quaternary carbons. The downfield signals at δ 79.01 and 124.34 were due to the hydroxy-bearing C-3 and vinylic C-12, respectively. The six methyl carbons resonated at δ 15.72, 17.48, 28.06, 23.53, 28.11 and 19.74 in the 13C-NMR spectra. The assignments to the various carbons in the molecule are presented in experimental section.
Two-dimensional NMR techniques such as COSY 45º, HOHAHA, HMQC and HMBC [11-13] were used to obtain more structural information. The one-bond 13C/1H couplings were determined by the HMQC spectrum see experimental section. The Homonuclear Hartmann Hahn spectrum (HOHAHA) [15,16,20- 23] recorded with mixing delay of 100ms showed that the C-3 methine proton is coupled with four protons i.e. with the C-2 and C-1 methylenic protons (δ1.23/1.25 and 1.61/1.81) respectively. The another two downfield exocyclic methylene protons appearing in the COSY-45º spectrum (4.54 and 4.65) not only displayed geminal coupling interactions but also gave strong cross peaks with the C-32 proton (δ 1.64/4.54, 4.65) in the same 2D experiment indicating the presence of the isopropene protons. These observations led to define the structure of 2 as 3β-hydroxy-24-nor-urs-4,12,28-triene triterpene.
3,28-β-diacetoxy-5-olea- triterpene (3), C33H52O4, was obtained as a white amorphous compound by column and preparative thin-layer chromatography of the ethanolic extract of A. scholaris flowers. The UV spectrum recorded in methanol showed terminal absorption only, while the infrared spectrum displayed absorptions for C=O of ester group (1735cm-1) and C = C (1630cm-1) groups [19,24-26]. The molecular composition was determined as C33H52O4 by high-resolution electron-impact mass measurements of the M+ (m/z 530).
The 1H-NMR spectrum (CDCl3, 500MHz) of 1 showed nine three-proton singlets at δ 2.13, 2.10, 1.15, 1.08, 0.97, 0.95, 0.94, 0.85 and 0.84 indicating the presence of nine tertiary methyls in the molecule as expected in a pentacyclic triterpenoidal skeleton. The downfield singlets at δ 2.10 and 2.13 were due to the C-31, C-33 acetate methyls. The downfield region of the spectrum also contained two signals i.e. a double doublet at 4.50 (dd, J3α,2α= 6.12, J3α,2β= 14.5Hz) and a broad double doublet at δ 5.62 (J6,7α = 4.0Hz, J6,7β= 12.5Hz), which could be assigned to the hydroxy-bearing C-3 methine and the vinylic C-6 protons, respectively [27]. A comparison of 1H-NMR chemical shifts of 1 with 3α-hydroxy-D-friedoolean-5-ene was also done [10].
The 13C-NMR spectra (broad-band decoupled and DEPT [10,12] CDCl3, 100MHz) of 3 exhibited signals for all 33 carbons and further supported the formula derived by mass spectrometric observations. DEPT spectra showed the presence of nine methyl, ten methylene, five methine and (by difference from the broadband decoupled spectrum) nine quaternary carbons. The downfield signals at δ 80.94, 121.70 and 170.96 were due to the acetate-bearing C-3, vinylic C-6 and acetate carbonyl C-28 respectively. The eight methyl carbons resonated at δ 31.87, 31.49, 29.20, 20.00, 19.39, 19.04, 18.04 and 12.05 in the 13C-NMR spectra. Two-dimensional NMR techniques such as COSY- 45º, HOHAHA [17,18,28], HMQC and HMBC [13] were used to obtain structural information.
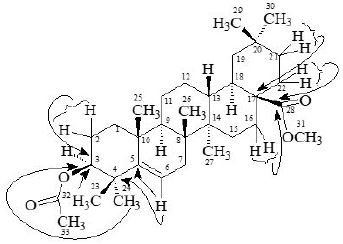
The Heteronuclear Multiple Quantum Coherence [13] (HMQC) spectrum of 3 displayed cross peaks between directly coupled carbon-proton pairs. HMQC interactions are presented in experimental section.
The Heteronuclear Multiple Bond Connectivity [13] (HMBC) spectrum was also very informative in determining the position of the double bond between C-5 and C-6, since the C-6 vinylic proton (δ 5.62) showed long-range interactions with C-4 (δ 40.45) and C-9 (δ 51.24). The C-3 proton (δ 4.50) showed coupling with C-5 (δ 140.78).

α-Amyrin acetate (4) M.P. 166-170 was isolated as a white amorphous solid from the pet. ether extract of A. scholaris by column and thin-layer chromatography. The high resolution electron-impact mass spectrum of 4 showed the molecular ion-peak at m/z 468.3708, which corresponded to the molecular formula C32H52O2. 13C-NMR chemical shifts of C-12 and C-13 at δ 124.33 and 139.63 respectively suggested that 4 belongs to Δ12-α amyrin series of triterpenoids with ester group in A/B rings.
Ursolic acid (5) was isolated as a greenish white powder from the pet. ether extract of A. scholaris by column and thin-layer chromatography. Compounds 4 and 5 were isolated for the first time from the flowers of this plant [4] and had not been previously isolated.
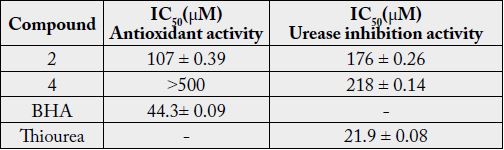
These compounds were analyzed for antioxidant and antiurease inhibition test. Compounds 2 and 4 were found to be antioxidant with IC50 107±0.39 and > 500μM respectively, while compounds 2 and 4 were also found to be urease inhibitors with IC50 175 ±0.26 and 218±0.14μM, respectively Table 1. The quantity of 1 and 3 was short, hence its activity could not be checked. BHA (IC50 = 44.3±0.09μM) and thiourea (IC50 = 21.0±0.08) were used as a positive control [29,30].
Experimental Section
The mass spectra were recorded on a Jeol HX-110 instrument. The 1H- and 13C-NMR spectra were recorded
in CDCl3 at 500, 400 and 125, 75MHz, respectively, on a Bruker AM-500, 400 NMR spectrometer. The UV
and IR spectra were recorded on Shimadzu UV-240 and JASCO A-320 spectrophotometers, respectively.
Optical rotations were measured on a polatronic D Polarimeter. The purity of the compounds was checked
on TLC (si-gel, Merck PF254, 0.25mm thickness). Melting points were determined in glass capillary tubes
using Buchi 535 and Gallenkamp 30/MF-370 melting point apparatus.
The flowers of Alstonia scholaris, (5kg) were collected from university campus Kashmir in October 2006. A
voucher specimen (AKUH # 58106) was deposited in the Herbarium of Department of Botany, University
of Azad Kashmir.
Air-dried flowers of Alstonia scholaris (5kg dry weight) were extracted with MeOH (50L). The MeOH
extract was concentrated to a gum (822gm), dissolved in distd. water and extracted thoroughly with pet.
ether (25L). The pet. ether soluble portion was evaporated under reduced pressure to yield a gum (66.92gm)
which was chromatographed on a si-gel column (Merck, 70-230 mesh, 2025.01gm). The elution of the
column was initiated with pet. ether. The combined column sub-fractions 1-8 (5.91gm) obtained by elution
with 5:95 ethyl acetate-pet. ether, which showed similar tlc behaviour upon spraying with ceric sulphate
reagent were combined and again subjected to CC using silica gel (type 60, 70-230 mesh, 200.10gm) and
column was eluted with pet.ether- ethyl acetate (99:1). The sub-fractions 6-30 (1.86gm), which showed
similar tlc behaviour were combined and further purified on preparative tlc plates using a solvent system of pet.ether- ethyl acetate (98:2) to afford pure compound 1 (19.5mg, 9.5 x 10-5% yield). The fractions
obtained on elution of the column with n-hexane-ethyl acetate (10:90) were checked on TLC. Fractions
7-18 showed similar behaviour on TLC were combined and further purified by preparative TLC (Merck
PF254 ,0.2mm) using CHCl3 as eluent to afford pure compound 2 (28mg, 1.4 x 10-4 % yield). Elution of the
major column which was loaded with 66.92g of pet.ether soluble material was eluted with 30% ethyl acetate
-pet.ether yielded an impure mixture (7.83gm). This mixture was again subjected to CC (silica gel, 70-230
mesh, 60.20gm). The sub-fractions 6-30 (1.86gm), which showed similar tlc behaviour were combined and
further purified on preparative tlc plates using a solvent system of pet.ether-ethyl acetate (90:10) to afford
pure compound 3 (19.5mg, 9.5 x 10-5% yield). The fractions obtained with 30: 70 ethyl acetate-pet.ether
yielded an impure compound 4, which was further purified by preparative tlc using a solvent system of pet.
ether- ethyl acetate (70:30) to obtain pure 4 (20mg, 10 x 10-4 % yield). Fractions 99-145 (1.91gm) obtained
by elution with pet. ether- ethyl acetate (35: 65, 500ml each) were collected and again subjected to CC
(70-230 mesh, 60.20gm). The sub-fractions 7-18 (0.92gm, 500ml each) obtained with 80: 20 hexane- ethyl
acetate showed similar behaviour on TLC (Ceric sulphate active) were combined and further purified on
preparative TLC (Merck PF254 ,0.2mm) using hexane- ethyl acetate 70:30 as eluent to obtain pure 5 (28mg,
1.4x 10-4 % yield, with Rf = 0.1).
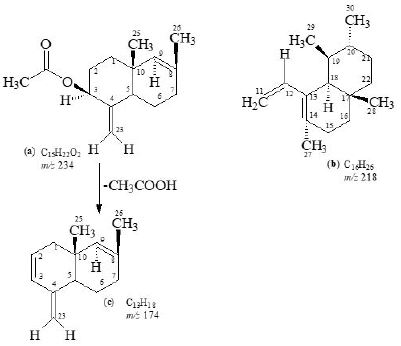
White solid substance, [α]29D = 40, UV: (MeOH) λmax nm: terminal absorbtion only, IR: (CHCl3) νmax cm-1, 1732 (C=O), 1630 (C=C), HREI MS: m/z 234.1618 (calcd. 234.1619), 218.2032 (calcd. 218.2034),
174.1406 (calcd.174.1408), F.D. 452, MS: m/z 452.3652 (calcd. 452.3654); 1H-NMR: (CDCl3, 500MHz),
13C-NMR (CDCl3, 125MHz) δ; data is presented in Tables-1, 2.
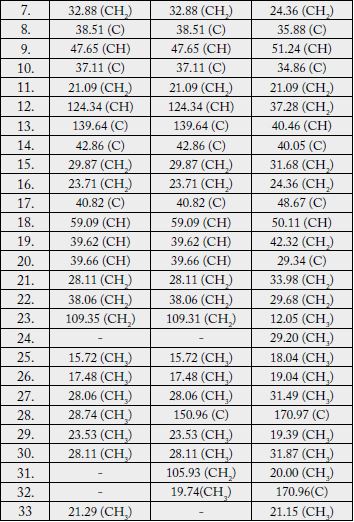
♣ Multiplicity assignments based on DEPT experiment # One-bond heteronuclear correlations determined by HMQC experiment
White amorphous substance, [α]29D = 40 (CHCl3), UV: (MeOH) λ
max nm: only terminal absorbtion, IR
(CHCl3) ν
max cm-1 hydroxyl (OH, 3415cm-1) and C = C (1630cm-1) groups, HREI MS: m/z 192.1513
(calcd. 192.1514, C13H20O) and m/z 244.2191 (calcd. 244.2190, C18H28), 174.1406 (calcd. 174.1408, C13H18 fragment c); F. D. 436; MS: m/z 436; 1H-NMR: (CDCl3, 500MHz), 13C-NMR (CD3OD, 100MHz) δ;
data is presented in Tables 1-2.
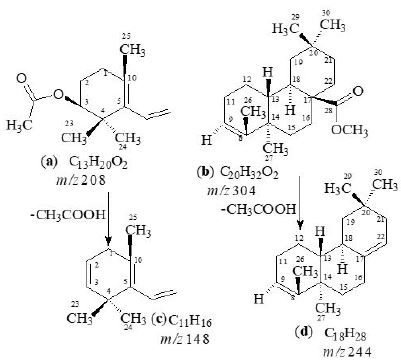
White amorphous compound; [α]D29 = 64.5º (CHCl3); UV: (MeOH) λmax nm: only terminal absorbtion,
IR (CHCl3) ν
max cm-1 (C=O, 1735cm-1), (C=C, 1630cm-1); EIMS: m/z 530, HREI MS: m/z 208.1462
(calcd. 208.1463), 304.2403 (calcd. 304.2402), 148.1252 (calcd. 148.1251) and 244.2191 (calcd. 244.2190);
1H-NMR (500MHz, CDCl3sub>); δ: 13C-NMR (100MHz, CD3OD) data is presented in Tables 1-2.
White amorphous solid; M. P. = 166-170ºC, [α]D29 = + 83.00º (CHCl3); UV λ
max (MeOH) nm, only terminal
absorption; IR (CHCl3) cm-1; 2850 (C-H), 1630 (C=C),1715 (C=O), 1136 and 1369 (C-O) cm-1 EI-MS
gave m/z at 469 (M• + H, C32H52O2), 454 (M• + H -CH3), 409 (42%), 394 (28%), 357 (15%), 298 (11%),
273 (39%), 267 (38%), 249 (68%), 232 (24%), 218 (100%), 203 (73%), 189 (75%), 175 (66%), 1H-NMR showed signals at δ
ppm= 0.78 (s, 1 x CH3), 0.86 (s, 2 x CH3), 0.87 (s, 2 x CH3), 0.97 (s, 1 x CH3), 1.00 (s,
1 x CH3), 1.06 (s, 1 x CH3), 2.02 (s, COCH3), 4.50 (m, 1-3β-H ), 5.11 (t, J= 3.6 Hz, C-12H), 13C-NMR
showed signals at 171.0, 139.71,124.41, 81.02, 59.16, 55.35, 47.73,42.82, 39.73, 39.72, 39.62, 38.56, 36.89,
34.82, 33.36, 31.32, 28.79, 28.45, 28.18, 26.68, 23.68, 23.44, 23.29, 23.29, 21.34, 18.32, 17.55, 16.94, 16.79,
15.77, 1H-NMR (CDCl3, 500MHz) δ: 13C-NMR (CDCl3, 125MHz) [31-33].
Greenish white powder, [α]D29 = 59 (c = 0.3, pyridine); M. P. 283-285 ºC, UV λmax (MeOH) nm; only
terminal absorption, EIMS m/z: 456.4 [M]+•, 438.4, 423.4, 300.3, 248.3, 203.2, 133.1; HRMS m/z: 456.3603
(C30H48O3), IR (CHCl3) νmax; 1H-NMR (CDCl3, 400MHz); 13C-NMR (CDCl3, 125MHz) [29].
Determination of DPPH Radical Scavenging Activity
The free radical scavenging activity was measured by 1,1-diphenyl-2-picryl-hydrazil (DPPH) [30]. The
solution of DPPH of 0.3mM was prepared in ethanol. Five microlitres of each sample of different concentration
(62.5μg - 500μg) was mixed with 95μl of DPPH solution in ethanol. The mixture was dispersed in 96 well
plate and incubated at 37ºC for 30min. The absorbance at 515nm was measured by microtitre plate reader
(Spectramax plus 384 Molecular Device, USA) and percent radical scavenging activity was determined in
comparison with the methanol treated control. BHA is used as standard.
DPPH scavenging effect (%) = Ac - As/Ac × 100
Where, Ac = Absorbance of Control (DMSO treated)
As = Absorbance of Sample
Reaction mixtures comprising 25μL of enzyme (Jack bean Urease) solution and 55μL of buffers containing
100mM urea were incubated with 5μL of test compounds (1mM concentration) at 30ºC for 15 min in 96-well plates. Urease activity was determined by measuring ammonia production using the indophenol
method as described by Weatherburn [34]. Briefly, 45μL each of phenol reagent (1% w/v phenol and 0.005%
w/v sodium nitroprusside) and 70μL of alkali reagent (0.5% w/v NaOH and 0.1% active chloride NaOCl)
were added to each well. The increasing absorbance at 630nm was measured after 50 min, using a microplate
reader (Molecular Device, USA). All reactions were performed in triplicate in a final volume of 200μL. The
results (change in absorbance per min) were processed by using SoftMax Pro software (Molecular Device,
USA). All the assays were performed at pH 8.2 (0.01M K2HPO4.3H2O, 1mM EDTA and 0.01M LiCl2).
Percentage inhibitions were calculated from the formula 100-(ODtestwell/ODcontrol) x100. Thiourea was
used as the standard inhibitor of urease.
Acknowledgement
The authors wish to thank the Ministry of Science and Technology, government of Pakistan for providing
the spectroscopic facilities for the current study.
Bibliography


Hi!
We're here to answer your questions!
Send us a message via Whatsapp, and we'll reply the moment we're available!