Biography
Interests
Rajneesh Verma1, Yogesh Shelar2*, Venkatesh, M. P.2 & Manoj Bansode3
1Stem Cells 21 co. Ltd, 2nd & 7th fl, Urbis bld, Aetas Residence, Soi Ruamrudee, Bangkok, Thailand
2JSS college of Pharmacy, JSS Academy of Higher Education and Research, SS Nagar, Mysuru, Karnataka
3Saiseva Biotech Pvt Ltd, CurecellsTM Cord Blood Bank, Kant Helix, Bhoir colony, Chinchwad, Pune, Maharashtra
*Correspondence to: Dr. Yogesh Kashinath Shelar, JSS college of Pharmacy, JSS Academy of Higher Education and Research, SS Nagar, Mysuru, Karnataka.
Copyright © 2019 Dr. Yogesh Kashinath Shelar, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
Supply chain management of vaccine is a crucial part in national immunization program. The vaccines used during this program should have to pass global standards for safety, efficacy and quality. To define these global standards the World Health Organization ( WHO) vaccines prequalification programme was started in 1987 [1]. Prequalification is the procurement term which helps to scrutinize the global public tender to limiting the best possible supplier for vaccine and immunization related devices.
This article describes the purpose of prequalification and the regulatory requirement for prequalification of vaccines in India. The article also describes the procedures for participation in prequalification programme by WHO along with requirements for vaccine production to the submission procedure to WHO. The article also includes the most common issues and difficulties faced by Indian manufacturers during the process of WHO-prequalification program.
In relation to this article, the Indian manufacturers may be able to understand the exact requirements of WHO-prequalification procedures and accordingly the objectives may be set by them for getting maximum benefits to the target populations of the country. Additionally, the manufacturer may refer annexure for getting the ready to use web links for referring different Guidelines for WHOprequalification procedures.
Introduction
In order to optimize use of health resources and improve health benefits to the mankind, WHO initiated prequalification programme which was started in 1987 to meet the global standards of quality, safety and efficacy of selected diagnostics, medicine, vaccines, immunization-related equipment and devices. Prequalification is the procurement term which helps to scrutinise the global public tender to limiting the best possible supplier. It worked as service provided by WHO to the supply division of the sister agency, United Nations Children’s Fund (UNICEF) and its prime purpose was to make sure that the vaccines purchased by UNICEF and other United Nations (UN) procurement agencies would be consistently safe and effective which meets global standards of quality to be used in the national vaccination programmes. The pilot project for testing of vaccines lots started with reviewing of the summary protocols and inspection of the vaccine manufacturing sites. The changing needs of UN agencies triggered the revision of procedures and every revision further endorsed and noted by the WHO Expert Committee on Biological Standardization (ECBS) prior to its implementation and publication as part of the WHO Technical Report Series (TRS) or as a Vaccines Department document [2,3].
Study Objective and Parameters
The objective of this article is to understand need of regulatory requirements for Vaccine approval by WHO
Prequalification procedure (PQP), to provide step wise process with specific details for WHO prequalification
procedure (PQP), difficulties faced by Indian applicants.
The following parameters were selected for the understanding and studying WHO prequalification procedure for vaccines:
1. The history and initial days of WHO prequalification
2. Purpose of WHO prequalification and NRA for regulatory requirements
3. Revised WHO PQ procedures
4. Steps of WHO PQ procedure
5. Difficulties/challenges faced and providing specific guidance for manufacturers in India
The schematic representation of methodological process is given as the flow chart in Figure 1.
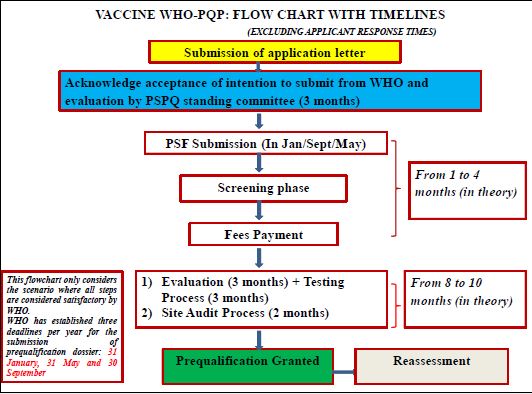
One of the major challenges being faced by WHO in the field of biological products was to make sure that vaccine used both nationally and globally would fulfill the appropriate quality norms. As the quality standards of vaccines was major issue because of the use of vaccines as “preventive medicine” in healthy populations, especially in many cases of newborns and healthy infants.
The Expanded Programme for Immunization (EPI) was started in 1974 and vaccines were supplied either by multinational manufacturers or national vaccine manufacturer. UN procurement agencies like UNICEF and the Pan American Health Organization Revolving Fund (PAHO RF) were procuring these vaccines with the conscious approach towards global standards of quality, safety and efficacy of the vaccines that were procured and distributed. In accordance with this one of the Immunization’s Newsletter started that “PAHO/ WHO screen manufacturers offering vaccines for EPI use and, where possible, review protocols of the specific lots submitted for sale” indicating the awareness for global standards. The PAHO Executive Committee encouraged all Member States to strengthen their respective testing laboratories for vaccine testing in its 84th meeting in June 1980. An agreement was established in 1987 between UNICEF and WHO to request its involvement in assessment of the acceptability, in principle, of vaccines for purchase through this agency [4].
The History and Initial Days of WHO Prequalification
WHO published its first set of requirements for prequalification in the year 1987, supplemented the following year by a modification applicable exclusively to BCG vaccine, because of the requirement as published in the TRS for annual clinical trials to establish the consistency of production of this vaccine. The early process focused on review of consistency of production was based on review of summary lot protocols and by quality control testing, but supplemented with clinical trials for BCG vaccine, and an inspection to the manufacturing site(s) [1].
In some cases, a small file was provided by manufacturers to give information about the production process, strain development and involvement of NCA in the oversight of the product. This started to be known as “the WHO prequalification procedure”.
The number of suppliers of a specific vaccine was limited to fewer than the total possible candidates by WHO on the basis of this evaluation process. This reduced population of suppliers of “prequalified vaccines” was further subject to a final “selection or qualification” that is carried out by UNICEF based on procurement principles and using a tender process that ends up in granting an award. The services were extended to the PAHO RF and later on considered as a standard service provided to any UN procuring agency including WHO HQ and all regional offices.
The objective of the WHO Prequalification ( WHO PQ) is to ensure over the quality of diagnostics,
medicines, vaccines and immunization-related equipment and devices in order to maintain their quality
standards. This will enable WHO to focus on high burden diseases for meeting the global standards of
Quality, Safety and Effectiveness for optimizing the use of health resources and improve health outcomes.
• Quality of diagnostics, medicines, vaccines and immunization-related equipment and devices needs to be
built & it should be reflected into the product, it can’t be tested in.
• It can provide quality products for UN procurement and country procurement.
• Lack of well-established drug regulatory systems (50% have varying capacity and level of development,
30% minimal or limited regulation).
• Increasing demand for generics, several players, and substandard products on the market.
• Deficiency in the quality of medicines/products can have serious consequences, which may turn into
ineffective treatment, drug resistance, side effects etc.
The amended procedure was reviewed and endorsed by the WHO Expert Committee on Biological
Standardization in October 2010 [6]. Each year, >2.5 billion doses of vaccines are being used globally to immunize children below 10 years old. About 65% of infants in the world are being vaccinated through
the WHO prequalified vaccines. The results of a WHO-PQ site audit led to a supply suspension of a
vaccine through United Nations systems in the year of 2012. The vaccine manufacturer actively quarantined
products in transit area and developed a corrective action plan (CAPA). Substitute sources of prequalified
vaccine were kept available to ensure continued supply.
• Eligibility for international, donor-sponsored tenders for Vaccines.
• Improved capacity to manufacture products for entry into stringently-regulated markets.
• Increased potential to compete successfully for contract manufacture for local markets.
• Faster registration.
• Improved image or brand.
• Status associated with producing quality-assured Vaccines.
• Enhanced image both externally and internally.
• Due to improved capacity utilization.
• Lower variable/commercial operating costs.
• Development of human resources for ensuring and managing quality manufacture.
• Capacity to ensure quality manufacture across range of products.
• New or increased capacity to meet stringent regulatory requirements.
In addition to securing a standard quality for vaccines for global supply, there was a need to ensure regulatory systems at national level to have the infrastructure and capacity to oversee the quality, safety and efficacy of products used within their territories. This was particularly important for countries with domestic production of vaccines and for those procuring vaccines directly through international bidding. WHO’s ECBS published in 1981 its first guideline on national control of vaccines, recommending the establishment of a “national control authority” (now called national regulatory authority) (NRA) for all countries, with responsibilities that may differ according to their capacity and need. A national control authority was recommended to be empowered to establish or to recognize requirements for acceptability of products, to establish standard preparations for biological testing, license manufacturers of biological products, and to establish the necessary infrastructure to implement their requirements. At this point, most of the emphasis was placed on the testing related activities. The Forty-fifth WHA in 1992, resolved that all vaccines used in national immunization programs should meet WHO requirements ( WHO 45.17), as a result reinforcing former procedures as a credible goal for the countries [3].
Therefore, this study was carried out to understand regulatory requirements for Vaccine approval by WHO Prequalification procedure (PQP), to provide step wise process with specific details for WHO prequalification procedure (PQP) and to discuss the difficulties faced by Indian applicants for WHO prequalification procedure (PQP).
Revised WHO PQ Procedures
An application letter has to be forwarded to Coordinator, QSSDOEMHP in WHO, vaccines PQ manager
and NRA, giving details and sites of manufacture, licensing status and the presentations proposed to UN
agencies procurement at any time with the expected date of file submission [7].
If require by manufacturer or WHO, the meeting will be held before the actual evaluation process starts.
Additional meetings may be held during assessment whenever needed.
Applicant has to prepare and submit 1 hard and 5 soft copies (on CD-RoM), in either MS Word or PDF
format, of a product summary file (PSF), which should be updated and written in English as per WHO
format mentioned as below:
• Chapter 1: General information
• Chapter 2: Personnel
• Chapter 3: Premises and equipment
• Chapter 4: Vaccine composition, presentations & schedules
• Chapter 5: Production
• Chapter 6: Quality control
• Chapter 7: Stability
• Chapter 8: Clinical trial data
• Chapter 9: Production & distribution data
• Chapter 10: Update on regulatory actions
However, now days, CTD format is accepted if: detailed cross-referencing is presented; and the required aspects not included in the CTD. Where the PSF cross-references to the CTD format, the documentation may be in electronic form only. Electronic documents should be in searchable text where possible.
WHO has given 3 deadlines in a year for submission of PSF: 31 January, 31 May and 30September. Applications should reach at WHO by the submission date, for consideration of review. Applications reached after the specified submission date are not considered for assessment.
The PSF contains the below elements:
Includes a brief information of the company, list of pharmaceuticals and non-pharmaceuticals manufacturing
activities as licences by NRA, description of the facility, number of employees engaged in activity, nonscientific
assistance, GMP compliance. It also includes description of quality management system, internal
and external auditing data, sources and quality of raw material procured for vaccine manufacturing.
It includes organizational chart that explains the areas like quality assurance, production, quality control,
identification of personnel and their responsibilities. The curriculum vitae for heads of production, quality
assurance and quality control, indicating educational and experience qualifications needed to be included.
Also includes outline arrangements for basic and continuing training and how records are maintained along
with the personal health records.
The premises and equipment will be examined during site audit. But, some prior information is required to
be submitted during filling PSF which includes floor planning describing quality control areas, air flow and
material flow, product flow, personnel flow, waste management area, classification of rooms, air handling
etc. Provide information about ventilation system, water management systems, maintenance system and
areas handling of hazardous materials. For products where a separate facility is required (e.g. Tetanus, Bacilli
Calmette-Guérin vaccine [BCG]), describe how separation is achieved. Describe qualification and validation
procedures, including computerized recording and controller systems. A description of the validation
master plan is required. Provide a brief description of the procedures for cleaning manufacturing areas and
equipment. For multipurpose areas, briefly describe the system for cleaning and testing between campaigns.
Mention the vaccine composition (including diluent). Describe the presentations made available to United
Nations agencies, including diluents (if applicable), combination products, forms, dose sizes, type of containers,
VVM type used, and descriptions of application devices (e.g. auto disable syringes) to be delivered with the
vaccine, if applicable. For both the final product and diluents, provide samples of primary container, labels,
boxes and package inserts to be used for United Nations agency supply (in English). French, Portuguese,
Russian and Spanish versions need to be made available before supply to United Nations agencies starts.
Include the calculated volume per dose in cm3 of the secondary packaging.
Provide the manufacturing formula for each antigen production, batch formula for bulk, estimated number
of vials and doses filled, batch numbering system for intermediates and final products. A detailed description
of manufacturing processes along with the product characterization has to be provided including history
of master cell banks/virus seeds. This to be included with manufacturing steps, area of procedures, IPQC
test performed, quality control tests, storage of intermediates and finished products as well. In case of
recombinant vaccines, a description of the construction and characterization of recombinant vector, as well
as source of it shall be provided. Also, describe the general policy for process validation. List the processvalidation
activities performed. Summarize arrangements for the handling of starting materials, packaging
materials, bulk and finished products, including sampling, quarantine, release and storage.
Provide quality control test along with appropriate characterization of starting material required as per
pharmacopoeia; list of raw materials meeting in-house specifications, including the tests performed and
specifications. Provide list of biological materials, BSE/SEs in final lot of vaccines and media with ingredients
along with specification and tests performed for identification. The quality control test list to be performed
on label and packing material(s) (primary and secondary and tertiary material). Describe the supplier’s
approval criteria of raw material and relevant certificates.
For intermediate products, list of the routine tests performed and specifications for intermediates as well
as list of all the validation activities including assays performed is required. Include copies of standard
operating procedures for critical quality control tests (uncontrolled copies or concise descriptions of the
method and re-test criteria are acceptable).
For finished product (including diluents), list of the routine tests and specifications for the final lot vaccine,
brief descriptions of the method and retest standard that are acceptable with standard operating procedures
in English, list of the assay validation activities performed and list of the final lots rejected during the last 2
years and the reasons for rejection are required.
Stability studies are expected to have been designed and conducted to meet WHO guidance. Information
on stability tests on intermediates includes containers to be used for intermediate products; fixing shelf-life
and storage conditions; quality control test methods, with specifications and basis for selection of tests for
stability; identification of production with batch numbers, batch size. Quantitative assay should be labelled
as numerical value rather than indicative of “pass” or “fail”. Stability information should include shelf life,
storage conditions, quality control test methods, stability profile determination and dates of manufacture of the lots, the lot numbers, the vial and dose size and the scale of production. The stability data should
be sufficient to justify the choice of VVM for the use with product (Vaccine vial monitor, Geneva, World
Health organization, 2006, accessed 20 February 2013). Include the data for diluents and reconstituted
vaccine for lyophilised vaccines. Explain the policy of assigning the date of manufacture of each component,
as well as the final product (e.g. combination vaccine) and diluents, as appropriate.”
Provide a simple list of all preclinical studies that were sponsored by the applicant in support of use in
clinical trials in humans, or for significant changes to manufacture or use. Include in the list any important
conclusions. For preclinical studies performed after initial licensure, indicate the reasons for these studies.
Any other particularly relevant reports regarding safety aspects, whether or not generated by the applicant,
should be provided.
Safety data should be submitted both in the case of the initial application for prequalification evaluation and
for reassessment purposes.
All clinical studies should be conducted as per ICH-GCP guidelines. The sponsor should provide a list of
all clinical trials performed in all countries that are relevant to the application for WHO prequalification.
These should include all studies sponsored by the applicant both before and at any time after initial licensure,
whether or not submitted previously to the NRA(s) where the product is licensed. A detailed summary
and interpretation of the safety and efficacy data obtained from the pre-licensure clinical studies and all
studies performed in the post-licensure period that support the current prescribing information is required
for the submission. The summary should pay particular attention to any data that are relevant to the use
of the product worldwide in WHO-recommended schedules (e.g. co administration of other vaccines).
In the absence of such data, the summary should provide a preclinical and/or clinical justification for the
extrapolation of the existing data to the likely circumstances of use after prequalification. This summary
should complement, and not replace, the summary written by an independent clinical expert and included
as Clinical expert report 12.
Whenever possible, the applicant should provide the clinical sections of the NRA assessment reports
from the country of origin and/or country where the vaccine is initially licensed. Assessment reports for
both initial licensure and any subsequent variations to the license for changes relevant to clinical data are requested. For each study the report should include final approved protocol, which should indicate the date
of protocol approval by the ethics committee and the NRA, evidence of registration in a clinical trial registry
that is included in the WHO International Clinical Trials Registry platform, compliance certificate with
GCP, brief summary, type of study, rationale, sites of study, date, number and age of subjects and statement
of conclusion on safety and immunogenicity.
Provide an independent clinical expert report on the clinical studies (evidence of Expertise and independence
should be provided). If the application for prequalification is based on the extrapolation of the existing
clinical data to the likely circumstances of use after prequalification, and if the data are old or there is a doubt
regarding the ethical or regulatory oversight of the trial, the report should discuss the degree of compliance
with WHO GCP recommendations and current guidance regarding preclinical and clinical trials with
vaccines.
Provide an outline of the post-marketing Pharmacovigilance plan for the product. Provide an outline of the
applicant’s procedures for the collection, onward notification and assessment of adverse events. Provide a
listing of all reported AEFIs for the vaccine in question in the last 5 years or since the last WHO reassessment.
As far as is possible from the reports received, applicants should list the type of reaction, lot number, date and
place of immunization, patient’s initials and age and, for immunization series, the dose number. A judgment
of seriousness and whether or not the event was expected (in the light of the prescribing information) should
be provided where this is possible from the information. An assessment of the relationship to the vaccine
made by a clinician and, where relevant, by the applicant company or its independent clinical expert, should
be included. Whenever periodic safety updated reports (PSURs) are available, these shall be submitted.
The PSURs should include information following the ICH format, from all geographical areas where the
vaccine is used, or the absence of such information should be defended.
For serious adverse events reported in the last 5 years, or as long as the vaccine has been marketed (when
shorter than 5 years), provide the fullest possible description of each case, including any information there
may be on investigations, actions, patient treatment and outcome. This information should be provided as
part of the PSUR.
Provide information on the quantity of finished product distributed domestically and exported in the
previous 3 years including distribution history which includes quantity of bulk vaccine destined for United
Nations agencies that have been supplied to contract fillers/packagers for finalization (list individually), list
of countries where product is licensed, summary of recording system for distribution, international packaging
shipment and validation protocols and reports for shipping. Describe the measures for complaints handling
and recalls including recall investigation system, procedures for corrective actions, and description of the
regulatory requirements in case of recalls [4].
Regulatory documents need to be provided which includes marketing authorizations, refusals/ withdrawal/
suspension, GMP certifications, clinical trial suspensions, dosage schedule modifications. Along with the
population that is going to be targeted and indication for those need to be submitted. It should also include
inspections conducted by NRA and foreign authorities within previous 2 years along with scope of inspection.
Upon submission, PSF will be screened for complete filling and compliances required. If not so, the
completed PSF is required to be submitted along with compliances and formats. Then manufacturer is
informed official to pay screening charges. At the time of screening, vaccine candidates must be in
compliance with the mandatory programmatic characteristics as defined by WHO’s Immunization Practices
Advisory Committee. Non compliance with mandatory characteristic will be result in rejection of PSF. If
the prequalification secretariat identifies a deviation from the critical characteristics or a unique, novel and
innovative characteristic, as defined by WHO a recommendation from the Programmatic Suitability for
Prequalification (PSPQ) Standing Committee is required. During its review and discussion, which will
lead to the formulation of recommendations, the PSPQ Standing Committee may engage in confidential
discussion with manufacturers and other technical experts approved by WHO and the manufacturer. Under
special circumstances, when there is limited access to a vaccine of public health importance, exceptional
consideration will be given regarding the suitability of vaccine candidates that are non-compliant with the
critical characteristics or that present with unique and innovative characteristics. This decision can be made
by the prequalification secretariat and will take into account the recommendations of the PSPQ Standing
Committee, public health needs and availability of alternative products. The screening process and review by
PSPQ should not be longer than 3 months. In case of rejection the recommendation for resubmission can
be given by PSPQ committee.
Prequalification procedure for vaccines evaluated by the EMA under article 58 of regulation (EC) No. 726/2004.
For vaccines (and all medicines) manufactured by European manufacturers (or at least those with a legal presence in the European Community) intended for exclusive use in markets outside the European Community, the EMA established mechanism (Article 58 of Regulation (EC) No. 726/2004) whereby the EMA may give a scientific opinion, in the context of cooperation with WHO.
After acceptance of PSF, WHO request manufacturer for submission of adequate samples of Vaccine (3 to
5 lots) with summary lot protocols for testing through contract laboratories. WHO conducts independent
and impartial quality control testing. Sometimes the bulk vaccines may be tested by WHO. The testing is
usually done in 3 months time. As per revised procedure, WHO may review of test results performed in
NCL of producing country.
WHO conducts site audit as a part of initial evaluation, follow up to corrective actions, for reassessment
purpose, as a part of complaints, AEFI or if the quality issue or problem is suspected wherein NRA of
country of origin may be invited for participation as Observers. WHO ask for market authorization, post
approval changes or variations, recalls or withdrawal of lots, etc. from manufacturer or NRA The audit report
is sent to manufacturer within 30 days of completion of audit.
An application letter is to be sent to the Coordinator, Quality, Safety and Standards, Department of
Essential Medicines and Health Products at WHO, with a copy to the vaccines prequalification manager
and the EMA, with details of the country and sites of manufacture and presentations offered. A statement
that the applicant acknowledges and agrees to the fact that the EMA will share the report of the CHMP
evaluators, inspection reports (manufacturing facilities and clinical trial sites) and test results, if available,
with the WHO prequalification team, as well as mutual immediate notification of quality or safety concerns
of the product. An electronic copy of the dossier submitted to the EMA for evaluation under Article 58.
Notification about the official medical control laboratory (OMCL) selected for any testing required by the
EMA for evaluation under Article 58 or for prequalification by WHO. After this screening fees need to be
paid as per WHO criteria.
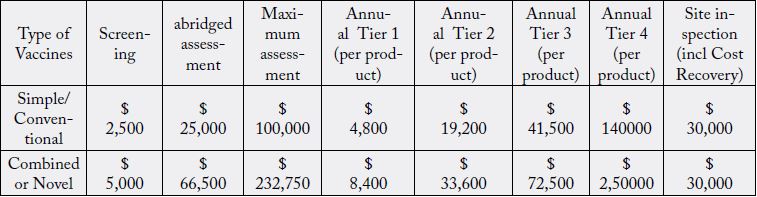
In the year 1999, WHO started to charge prequalification fees for vaccines which were motivated by the
need for financial sustainability for continuing the services and the fee structure was prepared considering
the following variables:
For vaccines:
• Nature of service: screening, evaluation, site inspections;
• Complexity of the product: simple or complex vaccine;
• Level of income generated by prequalification-enabled sales (sales to UN agencies and GAVI, only) to
determine which tier (from four levels) will be used to calculate the annual maintenance fee, payable per
product.
• Financially and administratively simple to carry out and observe.
WHO will carry out the evaluation the basis of EMA article 58 and annexed assessment report from
EMA/CHMP, a certificate of analysis of consistency lots by a qualified (OMCL) laboratory, reports from
relevant inspections (GMP, GCP and good laboratory practice) jointly agreed by WHO and the EMA
and performed. The essential criteria for review by WHO include confirmation about quality of vaccines
asper United Nation tender, stability data to assign VVM category, recommended immunization schedules,
packaging and shipment compliances and product characterization.
Once WHO considers that the process is complete, and if the outcome is satisfactory, WHO sends a letter
to UNICEF and other United Nations agencies, advising on the compliance of the vaccine with both the
WHO recommendations and the specifications of the relevant United Nations agency. The vaccine will
then be included in the WHO list of prequalified vaccines immediately after the letter to United Nations
agencies is sent.
After the prequalification of the product has been granted, follow-up activities to ensure continued
acceptability of the vaccine for supply through United Nations agencies will be performed according to the
general prequalification procedure which includes reassessments, evaluation of variations submitted by the
applicant.
This procedure is applicable for licensed vaccines which are used in routine immunization programme or in
emergency such as declaration of pandemicity, eradication, acute shortage etc. However, this is not applicable
for Novel vaccines or for vaccines recently introduced in routine immunization programmes.
After prequalification granted, the manufacturer should intimate post approval changes submitted to NRA
and even if not falls under post approval category that results in change in Safety and/or efficacy of the
vaccine or change the basis of approval of NRA periodically including annually.
Annual report should be submitted including details about post approval changes, NRA approval for the
post approval changes, ongoing stability programmes, production and distribution data, details of GMP
inspections, summary update on commitments given. The annual report should be first submission one year
after the date of prequalification.
It depends on the stringency of oversight exercised by NRA, interruptions to production/supply to UN
agencies, any failure, major variations from the last assessment or annual reporting.
Discussion
Supply chain management is critical in vaccine safety during national immunization programs. In this
respect, the role of the prequalification program is even more important in the post-prequalification phase
than before prequalification is granted. The continuing monitoring of quality and performance of prequalified
vaccines in use in the target countries is an important component of the activities of the program. This implies
testing of vaccine lots shipped to countries to ensure that the vaccines continue to meet the established
specifications (both WHO recommended and UN tender-related), review and approval of variations
introduced to the manufacturing process or quality control methods, monitoring and investigation of reports
of adverse reactions and of complaints.
The prequalification procedure was amended in 2010 and implemented from February 2012 aim at addressing:
• Reducing the period of prequalification
• Getting better help for complaints from the field and reports of AEFIs.
• Rising clearness and message to stakeholders and public for maximizing resources use [10].
With the introduction of a streamlined procedure stringent NRAs like Australia, Belgium, Canada, EMA, France, Italy and USA agreed to collaborate with WHO by providing the evaluation, inspection and QC testing reports and have established communication in case Quality, Safety or Efficacy of the prequalified vaccines are identified thereby reliance in job of NRAs will free capacity at the PQ secretariat However, it requires a closer monitoring because of scale up in manufacturing, improvement of product characteristics, changes in production or QC test methods [11].
Common Issues/Difficulties Faced by Indian Manufacturers for WHO-Prequalification Procedure
1. Many of the manufacturers in India are not aware about the WHO prequalification procedures or how to
approach WHO. Presently, only 8 out of 19 licensed manufacturers are having WHO prequalification for
their vaccines.
2. Lack of scientific approach/science during the development of candidate vaccine makes manufacturer
difficult to meet PSPQ criteria.
3. Non awareness of WHO priority list i.e. High, Medium, Low and no priority vaccine prior to submit
dossier for Pre-Qualification process: Manufacturers do not know about WHO priority for the vaccines and
hence most of the manufacturer fails to submit the application for WHP prequalification.
4. Lack of scientific, risk-based approach to respond to questions raised in the report of PSF.
5. Testing methods do not comply with WHO recommendations or are not fully developed and validated:
Sometimes the quality control testing methods are different in different pharmacopoeias and manufacturer
faces difficulties for meeting the requirements.
6. WHO GMP standards are not met as the vaccines are complex in nature and require more stringent
norms and conditions to meet as per the standards set.
7. Most of the manufacturers are having quality systems in place however they are lacking quality by design,
quality by culture systems and risk-based approach in maintaining the QMS systems.
8. Many of the manufacturers are still to develop the Pharmacovigilance systems. As it is one of the requirements
of prequalification and require a separate building system, infrastructure and resources, etc. It is expected
that manufacturers have to develop the risk management and Pharmacovigilance plan at the time of
market authorization. However, still manufacturers are finding difficulties for developing these tools.
9. As VVM is still not regulatory mandate requirement; many manufacturers are selling the vaccines without
VVM as it may add extra cost in the price of the vaccine.
10. VVM special studies are required to be conducted for suitability and presently there are very few manufacturers
of the VVM in the world.
11. Being thermo sensitive, vaccines are required to be shipped with special storage arrangements with ensuring
the cold chain maintenance throughout the sale/distribution/supply till to the level of patients.
12. Insufficient documentation proving that the trials were conducted after approval by ethics committees:
WHO requires statistical significant numbers for proving the safety and efficacy of the vaccines in
accordance with the disease prevalence. For getting approval, indigenous manufacturers are setting the aims
and objectives other than requirements of the WHO prequalification procedures.
13. Incomplete or no clinical development plan.
14. Poor quality trial reports with insufficient and/or contradictory information.
15. Clinical trial data produced with a different formulation than the one intended for marketing and for
distribution to UN agencies, without proper bridging.
16. Small database, mostly (but not only) from the safety perspective.
17. Immunogenicity determined with the use of commercial and/or in house non-validated tests.
18. Insufficient follow-up to determine duration of protection.
19. Lack of data in target population(s) that might receive the vaccine if prequalified, when such data are
needed.
20. Lack of data on co-administration of other vaccines.
21. Only immunological tests proposed when efficacy should be determined.
22. No post-marketing surveillance data from country of manufacture because the vaccine is produced for
export only.
23. Poor quality post-marketing surveillance data because of lack of adequate Pharmacovigilance system
(manufacturer’s and/or country NRA/MoH’s).
24. Manufacturers are not developing the post licensure safety study data of the vaccines as priority is being
given for marketing the vaccines at the earliest.
25. Further, manufacturers are unable to design the proper clinical trial data for co-administration with
other vaccines before the market authorization in the country.
Conclusion
With this article, the Indian manufacturers may be able to understand the exact requirements of WHO PQP
and accordingly the objectives may be set by them for getting maximum benefits to the target populations
of the country. Additionally, the manufacturer may refer guidance annexure for getting the ready to use web
links for referring different Guidelines for WHO PQP.
Limitation and Future Perspective
Currently the major limitation of prequalification program is a overall time of process and processing time
resources. The sample testing procedure during pre and post qualification is also need to be improved in the
prequalification process. This testing procedure need to be defined on global levels as the testing phase is
more important when the vaccines are introduced in immunization program. Also, the program needs to be
consistent in defining the GMP requirements.
In future the prequalification program needs to increase its efforts for achieving safe and effective quality products with reduction in overall prequalification procedure and approval time. This can be achieved by conducting parallel review process by NRA and WHO PQ team. There may be joint participation of WHO PQ inspector along with NRA inspection team while giving marketing authorization, approvals and its licensing of vaccines. Further, the prequalification regulatory framework needs to be built on national as well as regional levels for understanding and execution of WHO PQP.
Guidance (Annexure) for Industry for WHO Prequalification
1. Prequalification of Vaccines.
2. About WHO Prequalification of Vaccines.
3. Key Contacts
4. Procedure for assessing the acceptability, in principle, of vaccines for purchase by United Nation agencies, WHO TRS-978 Annex 6.
5. Assessing the programmatic suitability in principle of vaccine candidate for WHO prequalification (WHO/IVB/14.10). This document describes the WHO mandatory, critical and preferred characteristics of a candidate vaccine.
6. On WHO website for prequalification there is page containing the information and guidance documents for vaccine manufacturers. The following guidance can be found on this webpage:
Guidelines on the dossier preparation:
a. WHO web site for prequalification.
b. Vaccines prequalification priority list 2018-2020.
c. Vaccine Prequalification Dossier (Dec 2017).
d. Clinical considerations for evaluation of vaccines for prequalification.
e.https://www.who.int/biologicals/expert_committee/WHO_TRS_1004_web_Annex_9.pdf?ua=1
f. Guidelines on the international packaging and shipping of vaccines, WHO/IVB/05.23 -Vaccines vial
monitor.
g. Guidelines on Vaccine vial monitor (VVM).
7. Vaccine Prequalification including following:
a. A system for the prequalification of vaccines for UN supply
b. WHO list of prequalified vaccines for purchase by United Nations agencies
c. WHO list of contracted laboratories for vaccine prequalification
d. WHO Public Inspection Reports (WHOPIRs)
e. Issues relating to prequalified vaccines
f. WHO vaccines prequalification procedure
g. Assessing the programmatic suitability of vaccines candidates for WHO prequalification.
h. Collaborative procedure for registration of prequalified vaccines.
8. Model inserts for UNICEF packaging.
9. Environmental Monitoring of Clean Rooms in Vaccine Manufacturing Facilities.
10. The WHO Performance, Quality and Safety (PQS) catalogue:
11. WHO public inspection reports (PIRs):
12. List of WHO-contracted laboratories performing tests on behalf of the WHO vaccine prequalification programme.
13. Guidance On Variations To a Prequalified Vaccine.
14. Pharmacovigilance system (Related Publications)
• Reporting and learning systems for medication errors: the role of pharmacovigilance centres
• The use of the WHO-UMC system for standardized case causality assessment pdf, 152kb
• The Importance of Pharmacovigilance: safety monitoring of medicinal products
• Joining the WHO Programme for International Drug Monitoring pdf, 136kb
• Pharmacovigilance: ensuring the safe use of medicines
• Safety of Medicines in Public Health Programmes: pharmacovigilance an essential tool pdf, 592kb
15. Adverse Drug Reactions Monitoring.
16. Emergency Use Assessment and Listing (EUAL) mechanism.
17. Priority setting for WHO Prequalification.
Bibliography


Hi!
We're here to answer your questions!
Send us a message via Whatsapp, and we'll reply the moment we're available!