Biography
Interests
Rajneesh Verma1, Yogesh Shelar2, Venkatesh, M. P.2 & Manoj Bansode3*
1Stem Cells 21 co. Ltd, 2nd & 7th fl, Urbis bld, Aetas Residence, Soi Ruamrudee, Bangkok, Thailand
2JSS college of Pharmacy, JSS Academy of Higher Education and Research, SS Nagar, Mysuru, Karnataka
3Saiseva Biotech Pvt Ltd, CurecellsTM Cord Blood Bank, Kant Helix, Bhoir colony, Chinchwad, Pune, Maharashtra
*Correspondence to: Dr. Manoj Bansode, Saiseva Biotech Pvt Ltd, CurecellsTM Cord Blood Bank, Kant Helix, Bhoir colony, Chinchwad, Pune, Maharashtra.
Copyright © 2019 Dr. Manoj Bansode, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
Stem cell and cell derived products are included under the category of drug nowadays, as it is involve in treatment of several untreatable diseases. Therefore, in current era of modern medicine the stem cells need to pass through Food and Drug Administration (FDA) regulation of clinical trials to pass as a drug. International Society for Stem Cell Research (ISSCR) regulates clinical trials involved in stem cell research and its prospective human application. Currently every country including India has own regulation for stem cell research and are more or less similar to ICSSR. The role of National Apex Committee for Stem Cell Research and Therapy in India (NAC-SCRT) are clearly stated in NGSCR that approval for stem cell application in patients only within the confines of clinical trials and therapy based on Stem Cells and Stem Cell derived Products (SCCPs) should be informed in a proper format to the regulatory concern and done through the proper channel. This review article covers the requirement of laboratories and institutes which covered under NGSCR along with requirements for testing and evaluation of stem cells and cell based products in India. The review as a whole may not be comprehensive enough to include all the points in relation to the regulatory domain for approving Stem Cell Derived Product (SCDP) in our country but an attempt has been made to identify the important points keeping in mind that this domain will evolve and rules and regulations will change with the passage of time.
Abbreviation
ISSCR: International Society for Stem Cell Research
FDA: Food and Drug Administration
SCCPs: Stem Cells and Stem Cell Products
NGSCR: National Guidelines for Stem Cell Research
NAC-SCRT: National Apex Committee for Stem Cell Research and Therapy in India
SCDP: Stem Cell Derived Product
CLAA: Central Licensing Approving Authority,
IND: Investigational New Drug,
CRISPR-Cas9: Clustered regularly interspaced short palindromic repeats and CRISPR-associated protein 9.
iPSCs : Induced Pluripotent Stem Cells
MSCs : Mesenchymal Stem cells
HSCs: Hematopoietic stem cells
HLA: Human leukocyte antigen
GVHD: Graft versus Host Disease
AML: Acute myeloid leukemia
CML: Chronic myeloid leukemia
ALL: Acute lymphoblastic leukemia
HLA: Human leukocyte antigen
Introduction
Stem cells and cell-based products (SCCPs) have emerged with significant application in medical research
in the prevention or treatment of human diseases during the last few years. Stem cells can be defined as a
cell that has the ability to self-replicate unspecified period. The stem cells can be differentiated into different
kind of cell types under the specific conditions and signals and therefore have a potential to develop into
mature cells with specialized functions depending upon their origin and bio-plasticity. The application of
SCCPs is numerous in different clinical conditions such as cancer, ischemia, diabetes, Parkinson’s disease,
other neurological disorders and other diseases [1,2]. Currently, Human stem cell offers an extra-ordinary
approaches and opportunities in development of new medical agents that may act as regenerative medicine.
The current trend as a medicinal stem cell involves isolation, ex-vivo expansion and administration for the
therapy. Currently there are more than 100 diseases that can be treated with the help of stem cell therapy [3].
History and Evolution of Stem Cells and Cell-Based Products (Sscps)
Stem cell was first used as a drug to treat disease was during 1950s. During the 1950s the first implantation
with hematopoietic stem cells (HSCs) was carried out in 6 patients by intravenous administration. Despite
of absence of any side effects, the transplantation was failed in terms of non-survival of patients and nondetectable
marrow grafts. Started from this there is tremendous increase in homologous as well as autologous
marrow transplantation was observed from 1970 to 2007 [4]. Pre 2000s, all HSCs transplantation was
indicated for malignancies associated with haemopoietic system. But currently it is evolved for the treatment
of solid tumours [3,5,6]. In the initial part of 2000s with greater characterization of HSCs along with
Human leukocyte antigen (HLA) matching stem cells were commercialized in United State market and
introduce as a stem cells drug at 2010 with the name of Ducord and HemaCord. However, due to low HLA
matching ratio in the human population the HSC has evolved slowly in the market [3]. From 2012 onwards
Mesenchymal Stem cells (MSCs) are advanced as a stem cell drugs as they are having low immunogenicity
compared to HSCs and other cell-based products. MSCs also confirmed to exhibit strong modulation of
immunity and can regulate the immune system of host making them helpful in treatment [7-10]. Previously,
MSCs have been successfully transplanted in patients with the safety of HLA matching and absence of
immune suppression in patients developed them as a drug [11-13]. The first MSC based stem cell drug was
officially approved by FDA in Canada in 2012 which was indicated in the treatment of graft versus host
diseases relate to HSCT. In a pharmacology drug is a substance that is used to treat, cure, prevent or diagnose
a disease that causes physiological changes in body and helpful in management of disease when inhaled,
injected, consumed, absorbed via patch or by any other means. The substances that meet the indicated
criteria can be termed as a drug. Therefore, by definition stem cells can be enlisted into a drug category.
Currently there are two measure categories involved in stem cell drugs. First is HSCs based drugs which is indicated in treatment of Acute myeloid leukemia (AML), Thalassemia, Chronic myeloid leukemia (CML), Sickle cell anemia, Acute lymphoblastic leukemia (ALL), Aplastic anemia, Hodgkin’s lymphoma, Fanconi anemia, Non-Hodgkin’s lymphoma, Immune deficiency syndromes, Neuroblastoma, Inborn errors of metabolism, Ewing’s sarcoma, Autoimmune diseases, Multiple myeloma, Myelodysplastic syndromes, Gliomas, other solid tumors. Other is a MSCs based drugs which are utilized in when require immunomodulation, healing injury, promotion of tissue generation, stimulation of angiogenesis etc. MSCs are currently used in treatment of GvHD, degenerative osteoarthritis, repetitive trauma, Burger’s disease. The evolution of a concept in the realm of “modern medicine” changes with the passage of time. The evolution of the concept that stem cells should be considered as drugs per se, likewise changes with new findings. Findings on the Safety, Efficacy and large scale ex-vivo expansion of Pluripotent as for example Embryonic Stem Cells, or Multipotent as for example Mesenchymal Stromal Cells or iPSCs (Induced Pluripotent Stem Cells) are getting established every day.
To regulate the Stem Cell and Stem Cell-based products, the ISSCR’s (International Society for Stem Cell Research), came up with their guidelines in the year as recently as 2016 [14]. However these Guidelines couldn’t be accepted as the last word by FDAs, since it is an ever-evolving subject. ISSCR’s guideline was developed in 2016 by a group of scientists and ethicists from Asia, Europe, North America and Australia with through review from the data obtained from regulators, funding bodies, journal editors, patient advocates, researchers, stake holders, key opinion leaders in the public domain [14].
Definition of Stem Cell Derived Product (SCDP) [15]
• Stem Cell derived Products are used to refer to products, which are used in the form of drugs, intended to
be administered to a patient and that contains stem cells or is derived from stem cells.
• A drug which has been derived from processed cells including cell or tissue which has been processed by
means of substantial or more than minimal manipulation. (Substantial or more than minimal manipulation
means ex-vivo alteration in the cell population (T-Cell depletion, cancer cell depletion), expansion, which is
expected to result in alteration of function.
• With the objective of propagation and/or differentiation of a cell, cell activation and production of a cell
line.
• This includes, pharmaceutical or chemical or enzymatic treatment, altering a biological characteristic.
• Combining with a non-cellular component.
• And last but not the least, manipulation by genetic engineering, including application of CRISP-R, Cas 9
technique and gene modification.
Stem Cell Products from Indian Perspective
Stem cells or its derived products that are intended to administer to patients as in the form of drugs are
considered as Stem Cell derived Products. Stem Cell derived Products may be sampled and sent for quality
control testing and if required necessary legal procedures as per the provisions of Drugs and Cosmetics Act
can be taken by the regulator. The stem cells shall pass the proof of concept, animal toxicity, and science
based approach for the process of approval or licensing. All stem cell derived products are mandated under
drugs and cosmetic act.
Stem Cell Therapies (SCT) from Indian Perspective
When the stem cells and stem cell derived products as a part of all invasive procedure by physicians, doctors
and clinician as a part of standard care treatment then it is referred as a Stem cell therapy. Most widely
used stem cell therapy includes bone marrow transplantation and therapies derived from umbilical cord
blood. These therapies are procedure by medical practitioners under the medical ethics as it is difficult to
make provisions for sampling, testing and other legal procedures for stem cells therapy under Drugs and
Cosmetics Act. Currently stem cell therapies are regulated by ICMR, NGSCR and Indian Medical Council
Act [16].
The difference between stem cells products and cell therapies indicated in Table 1.

Stem Cell Research Regulations in Different Countries
Different countries have attempted to regulate SCDP and have come up with different guidelines. The
leading countries who have extensively worked on stem cell research and development and contributed in
the development of products are USA, Japan, UK, Korea, Australia, China, etc. The comparisons of current
regulations for stem cells are indicated in Tables 2, 3 and 4.
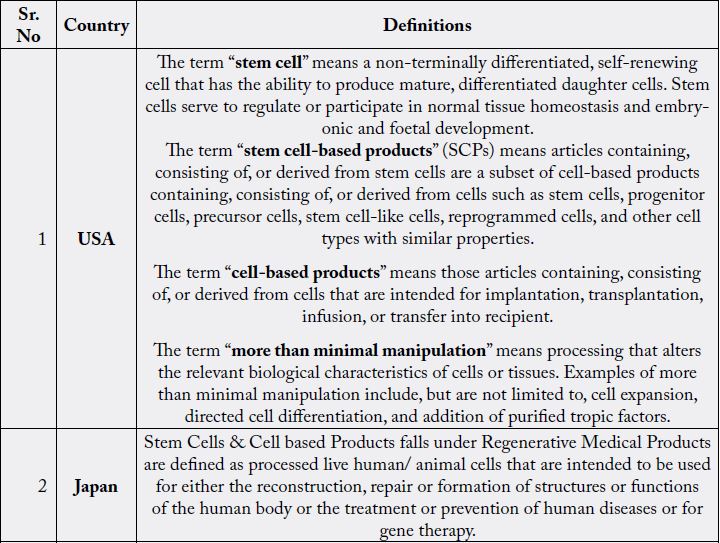

In USA stem cells and cell-based products are divided into two categories i.e. 351 and 361. The product
category 351 involves cells that are more than minimally manipulated and to cells that are used in a nonhomologous
manner. The USFDA developed a regulatory framework for controlling cell and tissue based
products based on three general areas which include prevention of use of contaminated tissues or samples,
prevention of improper handling or processing which may lead to damage or contamination of cells and
clinical safety of all the tissue or cell that are processed for functional use [18].
FDA introduced three types of regulatory exceptions which are granted to cells and tissue products [19]. This includes speeding up the transitions from preclinical testing and accelerated authorisation in clinical trials that involves serious patients with considerate of low life. It also includes the rapid testing of breakthrough therapies that are potential in the treatment of serious threatening diseases. In addition to these FDA introduced priority review procedures which aims towards the decreasing time frame required for evaluation process after completion of clinical trial. The regulatory exception also includes expanded access program which provides patients access to the parallel treatments which includes the biological drug products [20].
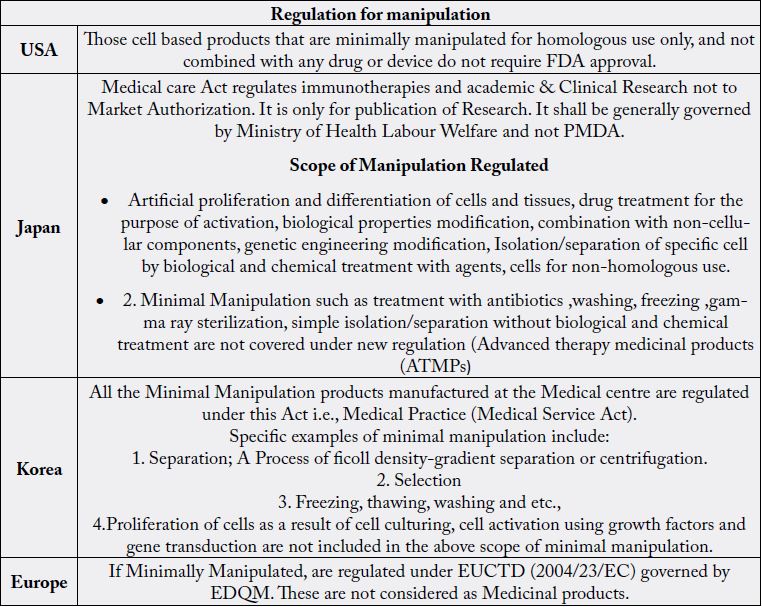
The “361” category regulates use of minimally manipulated stem cells and are functional for homologous use. These products can be used in patients under compliance with US human tissue regulation [21]. The majority of self-classified ‘361’ product are actually unproven ‘351’ products which are offered illegally to the patients. To overcome this USFDA made more stringent regulatory approach which involves these products need to have a USFDA pre-market approval. Also, these products should follow approval process through the proper pipeline involving the preclinical studies, investigational new drug (IND) application and clinical trials. This is having similar regulatory exceptions as ‘351’.
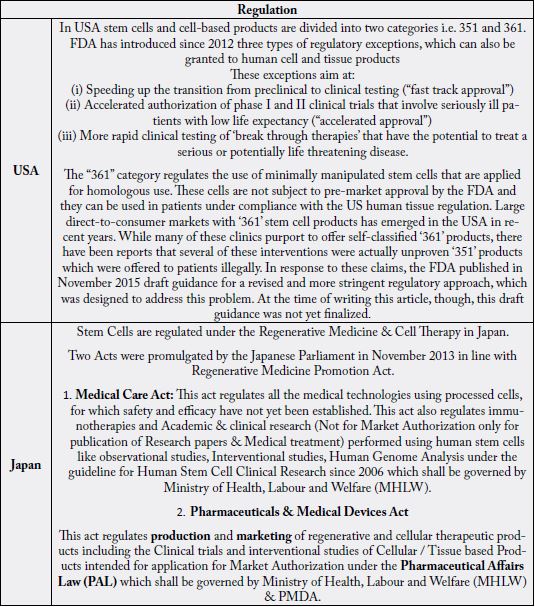
As per European Union stem cells are the one those have self-renewing capacity i.e., capability to generate
daughter cells and multi-lineage differentiation capacity. Stem Cells are capable of proliferation as stem
cells in an undifferentiated form. This includes embryonic stem cells (hESCs) derived from blastocysts;
Haematopoietic progenitor stem cells (HSCs); Mesenchymal Stromal/stem cells (MSCs); Induced
Pluripotent Stem Cells (iPSCs).
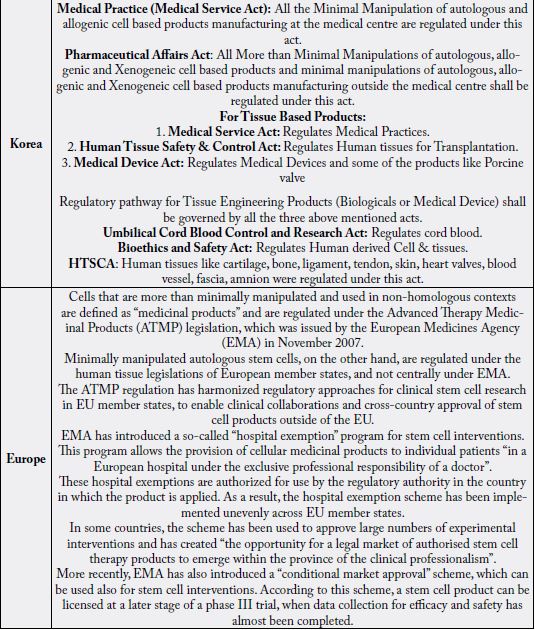
Regulatory arrangements for stem cell treatments in the EU are similar to the USFDA. Cells that are more than minimally manipulated and used in non-homologous contexts are defined as “medicinal products” and are regulated under the Advanced Therapy Medicinal Products (ATMP) legislation issued by the European Medicines Agency (EMA). While in case of minimum manipulated autologous stem cells are not covered under EMA, but under human tissue legislations of European member states. As like that of USFDA, the EMA also require the systematic clinical studies as though there is no large scale market emerged in minimally manipulated stem cells [22]. As similar to USFDA, the EMA has introduced a “compassionate use” program, which allows access to new drugs and biological products (including stem cell products) outside of premarket clinical trials.
Recently, EMA introduced a scheme named as conditional market approval. As per this scheme, a stem cell product can be licensed at a later stage of a phase III trial, when data collection for efficacy and safety has almost been completed. If Minimally Manipulated, are regulated under EUCTD (2004/23/EC) governed by EDQM. These are not considered as Medicinal products.
Regenerative Medicine Promotion Act (RMPA) was passed by Amended Pharmaceutical Affairs Law,
which went into effect in November 2014. As per RMPA, stem cell and cell-based products are covered
under the Regenerative Medical Products and are defined as processed live human/ animal cells that are
intended to be used for either the reconstruction, repair or formation of structures or functions of the human
body or the treatment or prevention of human diseases or for gene therapy. The RMPA also regulates the scope of manipulation. This includes artificial manipulation and minimal manipulation but blood transfusion
(blood Products), Hematopoietic stem cell transplantation, Assisted Reproductive Technology, except those
derived from genetic engineering, Human induced pluripotent stem cells (HiPSC), are excluded from the
scope. RMPA allowed for conditional market approval for stem cell products after early phase clinical trials.
Conditional approval can occur after positive clinical data from as few as ten patients, provided these firstin-
human-trials demonstrate that the tested cell products are safe and “likely to predict efficacy”. Costs for
these experimental treatments are split between the state and patients at a ratio of 70:30.
1. Medical Care Act: This act regulates all the medical technologies using processed cells, for which safety and efficacy have not yet been established. This act also regulates immunotherapies and Academic& clinical research (Not for Market Authorization only for publication of Research papers & Medical treatment) performed using human stem cells like observational studies, Interventional studies, Human Genome Analysis under the guideline for Human Stem Cell Clinical Research since 2006 which shall be governed by Ministry of Health, Labour and Welfare (MHLW).
2. Pharmaceuticals & Medical Devices Act: This act regulates production and marketing of regenerative and cellular therapeutic products including the Clinical trials and interventional studies of Cellular / Tissue based Products intended for application for Market Authorization under the Pharmaceutical Affairs Law (PAL) which shall be governed by Ministry of Health, Labour and Welfare (MHLW) & PMDA. A separate category and definition of Regenerative Medical Products has been made under PMDA, as it is difficult to gather and evaluate the data for efficacy of Regenerative Medical Products in a short time due to heterogeneity of cells. An expedited approval system for Regenerative Medical Products has been followed, After the safety is confirmed and results predict likely efficacy, the product will be given conditional time limited Market Authorization in order to enable timely provision of the products to the patients. To approve products based on the limited data, such as surrogate endpoints in exploratory study shall be used Similarity to accelerated approval of USFDA. The product has an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit.
All the Minimal Manipulation of autologous and allogenic cell-based products manufacturing at the medical
centre are regulated under this act.
All More than Minimal Manipulations of autologous, allogenic and Xenogeneic cell-based products and
minimal manipulations of autologous, allogenic and Xenogeneic cell-based products manufacturing outside
the medical centre shall be regulated under this act.
For Tissue Based Products the regulatory pathways are
4. Medical Service Act: Regulates Medical Practices.
5. Human Tissue Safety & Control Act: Regulates Human tissues for Transplantation.
6. Medical Device Act: Regulates Medical Devices and some of the products like Porcine valve
All the Minimal Manipulation products manufactured at the Medical center are regulated under this Act i.e., Medical Practice (Medical Service Act).
The clinical use of stem cells is regulated from the Argentinean Ministry of Health under Ministerial
Resolution No. 610/2007.
This resolution states that the use of human cells falls under the authority of the Unique Central Institute for Ablation and Implantation (INCUCAI). By falling under the authority of INCUCAI, stem cell interventions are not governed as a medical product (as in the EU, India and the USA) but as a medical procedure, which are managed by the Argentinean Transplant Act. With the exception of haematopoietic cell transplants from human bone marrow, all types of stem cells are considered experimental and require evaluation of safety and efficacy through clinical research.
The regulatory framework for stem cell research is prohibited due to two different reasons that is
Brazilian constitution prohibits the commercialization of human body materials, including human cells and their derivatives.
Salient Features of Guidelines of Stem Cell Research in India
It is universally accepted that stem cells are required to undergo processing by ex-vivo or in- vitro methods
to get a large number. In case of getting these cells to be functional minimum manipulation of the stem
cell is necessary. The current challenge in stem cell research is to evaluate the potency of stem cell through
animal models [24].
Accordingly novel surrogate assays and “ORGANOID” systems may be required for this purpose. It is obligatory that the Stem Cell or Stem Cell-derived Products (SCDP) are processed in CDSCO approved Good Manufacturing Practice (GMP) approved centres throughout the country. The discipline of “Quality Control and Quality Assurance” (QC/QA) for product development, inclusive of manufacturing protocols and cell processing is essential and should be in line with the regulatory mandates of the Schedule M of Drug and Cosmetic Act, 1940 and its contents [25].
• SCCDP (Stem Cells and Stem Cell derived Products) should have proper label before they are released.
• SCCDP intended for human application (Strictly Investigational)/as a part of clinical trial should be in
strict adherence to QC/QA as defined in Annexure VI (NGCSR-2017).
• These include “CELL VIABILITY”, “Final Cell Population (Checked for CD markers by Flow Cytometric
Techniques), “STABILITY” and “REQUIREMENTS and RELEASE”.
• It is required that SCCDP is sufficiently stable for the “duration” of the study.
• All procedures shall be formulated and clearly stated in the form of SOPs (Standard of Practices) and
strictly adhered to under all circumstances, so as to ensure, “REPRODUCIBILITY” of well characterized
“CLINICAL GRADE” cells that meet the desired criteria of the following: “IDENTITY”,
• And last but not the least, the “GROUNDWORK FACILITY” should be duly approved by “CDSCO”
and the same should grant the approval after receiving the CMC (Chemistry, Manufacturing and Control)
documents.
• At the present juncture there is a of “DEARTH” of solid “SCIENTIFIC” proof of principle studies to substantiate
the clinical EFFICACY of STEM CELLS in pathologic states, other than the use of the same in
Hematopoietic Stem Cell Transplantation (HSCT) for “APPROVED” indications as reflected in Annexure
III of NGSCR, 2017 and rest of the therapies come under developing or research categories.
• So there is a complete “Sanction”/ “Prohibition” with regard to the “COMMERCIAL” use of stem cells as
examples of therapy.
• It must be categorically emphasized that not a single stem cell administration is outside the purview of
clinical trials.
• The experimental or investigational protocols within the purview of clinical trials should be designed with
care and should have “PRIMARY” and “SECONDARY” end points.
• The follow-up period should be minimum of “TWO” years OR more depending on the “CATEGORY”
and “ORIGIN” of stem cells used the intended clinical application as well as, the “AGE” and “GENDER”
of the patient treated in the clinical.
• It is of paramount importance that the “STAKE HOLDERRS” involved in STEM CELL and STEM
CELL-DERIVED product research and application are fully aware of current regulation and best international
practices in the field, inclusive of existing cGMP and Good Clinical Practice (GCP) arbitrations.
• Moreover an enrolled human participant in a clinical trial should not be charged, unless approved by the
regulatory authorities, for any procedures, in relation to the trial including hospital stay, and clinical tests
performed on him or her during their stay.
• Stem Cells and Stem Cells derived products are associated with unique ethical considerations and obligations.
• Legal and Social concerns that demands additional oversights and expertise from well-trained ethicists,
scientists, seems particularly important.
• A distinctive mechanism for review and monitoring is obligatory at both institutional and national levels.
• A National Apex Committee for Stem Cell Research (NAC-SCRT) has been established, to oversee research
activities, and putting into place guidelines for basic and clinical research at the National Level.
• On the other hand the “Institutional Committee for Stem Cell Research(IC-SCR)’s duty is to primarily
“Approve” and monitor Stem Cell Research (Basic and Clinical) at the institutional level.
• The responsibilities, functions and attributes of both IC-SCR are given in annexure I of the recent of
NGSCR-2017.
• These “Watch-Dog” committees shall ensure that they review, approve and monitor processes and protocols
of stem cell research being carried out in accredited Laboratories in the national level, which are in
compliance with the national guidelines.
• It is mandatory for all institutes, stakeholders, laboratories, who are involved in Stem Cell Research to
establish an IC-SCR and register the same with NAC-SCRT.
Stem cells, whether autologous or allogenic demands variable degree of ex vivo or in vitro processing/ manipulation before their use of clinical application/transplantation and translational research.
• This may carry the risk of “Contamination” and may also lead to alteration in their properties which is in variance with the methods applied for manipulation.
• All laboratory procedures should be carried out under aseptic conditions in CDSCO certified GMP (schedule M) and GLP (Schedule L1) facility for human applications.
• For pre-clinical studies on animals the laboratory should have GLP certification from the Department of Science and Technology (DST).
1) Processing includes simple isolation/separation, washing, centrifugation and suspension in culture
medium/reagents, cutting, grinding, shaping, and overnight culturing without biological and chemical
treatment, de-cellularization.
2) Clinical applications, using such cells require the IC-SCR, IEC and CDSCO approvals if these are
meant for homologous use.
3) If the minimally manipulated cells are to be used for non-homologous purposes, CDSCO’s approval is a
must from those of the NAC-SCRT, IC-SCR and IEC before initiating any clinical application.
4) If the cells/tissues are removed and implanted into the same individual during the same surgical procedure
within a single operation, it should not undergo processing steps beyond centrifugation/rinsing/cleaning
and sizing.
1) This can be categorized as ex vivo processing/manipulation via which the cells are permanently altered
(augmentation or decrease in specific subsets), expansion, cryopreservation or cytokine-based activation,
albeit one that is not expected to the cellular characteristics and functioning.
2) Clinical trials using cells that have undergone more than minimal manipulation need approval from
CDSCO after procuring approval from CDSCO, IC-SCR and IEC.
3) Clinical Trial using SVF or Stromal-Vascular Fraction from Adipocytes requires approval by IC-SCR,
IEC and CDSCO.
1) This is in relation to “GENETIC” and “EPIGENETIC” modification of stem cells, transient or permanent,
or of cells propagated in culture leading to alteration, not only in numbers, but also biological characteristics
and function.
2) This includes trans-differentiation, transduction or transfection by lenti/retro viruses as well as other gene
delivery modes with the soul intention of having specific selection & clonal expansion of a specific cell of
interest.
3) Use of stem cells which have been subjected to maximal or major manipulation definitely requires approval
of CDSCO after obtaining the clearance from NAC-SCRT through IC-SCR and IEC before moving on
to clinical applications.
Types of Research [27]
• The research has been delineated into three basic types, based on ‘ethical’, and or ‘safety’ concerns, regarding
the origin of stem cells and levels of manipulation.
• The latter warrants surplus reviewing and monitoring as per the existing norms.
• They are further categorized into “PERMISSIVE”, “RESTRICTIVE” and “PROHIBITIVE” areas.
1. In vitro or ex vivo studies using Stem Cells or Stem Cell-derived products approval has to be obtained
from IC-SCR and IEC.
2. Establishment of novel human embryonic stem cell lines from spare embryos or iPSC lines from fetal/
adult somatic cells or somatic stem cells from fetal or adult tissues with prior approval of the IC-SCR and
IEC.
3. If the tissue is procured from hospital/clinic/entity, other than the concern of the researcher, then the IEC
clearance is obligatory.
4. In vitro studies using established cell lines can be carried out with prior permission from the IC-SCR.
5. In vivo studies in experimental animals excepting primates, using established cell lines from any type
of human pluripotent stem cells, for instance iPSCs, ESCs, as well as their differentiated cells and human
somatic stem cells, (viz., fetal, neo-natal or adult) from any tissue, are allowed only when IC-SCR and
Institutional Animal Ethics Committee (IAEC) comes into the picture.
Inclusive of both basic, pre-clinical and translational research domains, demanding additional elements of
supervision or monitoring due to contentious issues.
• Creation of human pre-implantation embryos by IVF technology, ICSI (Intra-Cytoplasmic Sperm Cell Injection), Somatic Cell Nuclear Transfer (SCNT), etc, with the sole objective of making cell lines. The researcher/investigator needs to provide appropriate reasoning paying attention to the following points:
1. The proposed research cannot be carried out with existing ESC lines, or those that can be derived from
surplus embryos.
2. Minimum number of embryos or blastocysts required for such research must be categorically stated in
writing.
3. Research teams involved in ESC research should be having the expertise and knowhow to do the same in
GLP certified and NABL accredited laboratories.
• Clinical trials using stem cells of any type, source and level of manipulation for homologous/nonhomologous transplantation in indications other than mentioned in annexure III, can only be done with prior permission from IC-SCR, IEC and CDSCO.
• Trials should be done; using clinical grade cells in GLP and GMP compliant facilities.
• Foreign nationals or multinationals, employing Stem Cell or Stem Cell-based products outside the jurisprudence of India, wanting to do clinical trials in India should take clearance certificates and approval, from their country of origin, to undergo clinical trials and ensure that they are approved to carry out such trials by IC-SCR and IEC.
• All international collaborations require approvals from the respective countries from which they are coming, approval from their funding agencies followed by approval from Health Ministries Screening Committee, in line with the Government of India Guidelines (Available at http://icmr.nic.in/guide.htm ).
• Import of any type of stem cells and or stem cell-derived products/ derivatives demands license from CDSCO as per the existing guidelines. These should have prior clearances from the regulatory authorities of the respective countries.
• Studies on Chimeras where stem cells from two or more species are mixed together at any stage of early development (embryonic or fetal) for having a clear understanding patterns and pathways sponsoring development and differentiation would also require prior approval of NAC-SCRT after clearance has been granted by IEC and IC-SCR.
• GENOME MODIFICATION including “Gene Editing” (for example by CRISPR-Cas9 technology) of Stem Cells, Germ Line stem cells or gamete and human embryos is restricted only to ex vivo and in vitro studies.
• This area of research will be required to be thoroughly reviewed y Review committee on Genetic Manipulation (RCGM).
• In the present scenario with respect to Scientific Knowledge and understanding, stem cell research in the
following areas is prohibited.
• Research Related to Germ Line Gene Therapy and Reproductive Cloning.
• In vitro culture of Intact Human Embryos, regardless of the protocol of their derivation, beyond 14 days
of Fertilization or generation of Primitive Streak or whichever is earlier.
• Clinical Trials involving “XENOGENIC” cells.
• Any clinical research on human- xenogenic hybrids.
• Use of genome-modified human embryos, germ-line stem cells and gametes for developmental propagation.
• Research in the domain of implantation of human embryos (generated by any means) after in vitro
manipulation at any stage of development in uterus humans or other species of primates.
• Breeding of animals in which any type of human stem cells have been introduced at any stage of development,
and may be considered to be incorporated in the germ line of the animal in future.
Guidelines for Clinical Trials [27,29]
The guidelines associated with clinical trials with STEM CELLS should be in strict compliance with “NEW
DRUGS & CLINICAL TRIAL RULES 2019” of Drugs and Cosmetics Act 1940 and rules therein along
with GCP (Good Clinical Practice) guidelines of CDSCO (available at http://www.cdsco.nic.in/html/
GCP1.html) and ICMR ethical guidelines for Biomedical research incorporating humans (available at http://www.icmr.nic.in/ethical_ guidelines.pdf).
The researcher should strictly adhere to the Clinical Trial template for protocol in accordance with the given format (Annexure II) and submit the application to CDSCO in the form “CT-04”. Only institutions possessing their IC-SCR, registered with NAC-SCRT and IEC registered with CDSCO are allowed to undertake stem cell research.
Overview of Regulatory Requirements of Stem Cells and Stem Cells Derived Products in India [15]
• Central Licencing Approving Authority (CLAA) under Central Drugs StandardControl Organisation
(CDSCO) is empowered to regulate SCDP in India under Rules 21 (b) of Drugs &Cosmetics Rules.
•SCDPs are new drugs and approved as per New Drugs and Clinical Trial Rules 2019 under Drugs &Cosmetics Act, 1940 & Rules there under.
•Any breakthrough drug is regulated under the said Act with provision of exemption of drug approval
process like to conduct Phase-I & II together, issuance of conditional approval with condition to conduct
structured Phase-IV, PMS etc to find out residual confirmation of safety & efficacy (if required).
•The detailed regulatory pathways prepared by the committee formed by CLAA with the Experts in the
field having clinicians, researcher, academia, scientist, regulators, prepared by forming the committee.
•The regulatory requirements include:
(1) Application for permission to import for the sale or distribution
(2) To undertake clinical trials
(3) Manufacture of stem cell and cell / tissue derived products for sale or distribution.
The applicant will submit their application (for Import / Manufacturing) as per the format specified to the
CDSCO, where the application shall be scrutinized and technical reviewed.
If required onsite inspection shall be performed in this regard for verification of compliance. The Proposal shall be deliberated in the CBBTDEC / Apex / Technical Committee for the expert opinion on the proposal. Based on the recommendations of committee, NOC is granted to the applicant by the CDSCO.
The applicant is required to obtain Clinical Trial Permission in form CT-06(application shall be made in
CT-04), Market Authorization from CLAA in Form CT-23, before obtaining manufacturing license in
Form 28 from State Licensing Authority.
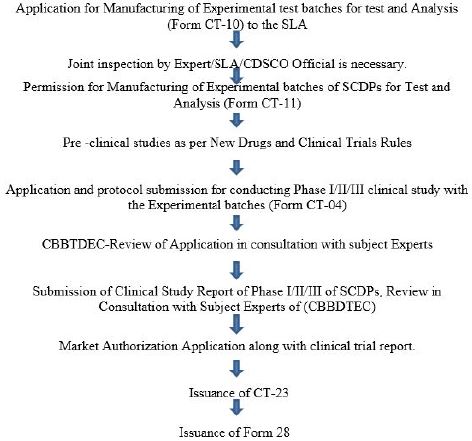

Committee’s Involved in Approval for Import / Marketing Authorization of the Stem Cell Derived Products
CBBTDEC (Cellular Biology Based Therapeutic Drugs Evaluation committee) is constituted by the
Government of India, Ministry of Health & Family Welfare under the Chairmanship of Secretary,
Department of Health Research to advise CLAA in matter pertaining to regulatory pathway leading to the
approval of clinical trials and marketing authorization for the Stem Cell and Stem cell derived products.
Technical Committee is constituted by the Government of India, Ministry of Health & Family Welfare
under the Chairmanship of Director General of Health Sciences to supervise and monitor the conduct of
clinical trials in the country. The objective of the technical committee is to oversee the conduct of clinical
trials and give its recommendations to the Apex committee for taking further appropriate action.
It is constituted by the Government of India, Ministry of Health & Family Welfare under the Chairmanship
of Secretary, Department of Health & Family Welfare. Joint Secretary, Ministry of Health & Family Welfare,
as Member Secretary to supervise the clinical trials on new chemical entities conducted in the country.
The objective of the Apex committee is to consider the recommendations of the Technical Committee on the approvals of Clinical trials and other related issues for appropriate direction in the matter.
•Submission of clinical trial application for Evaluating Safety and Efficacy.
•Requirements for permission of New Drugs Approval
•Post Approval Change in biological products:
•Quality safety and Efficacy Documents
•Preparation of the Quality Information for Drugs submission for New Drugs Approval.
Features of New Drugs and Clinical Trial Rules 2019
(i) A drug, including active pharmaceutical ingredient or phyto-pharmaceutical drug, which has not been
used in the country to any significant extent, except in accordance with the provisions of the Act and the
rules made hereunder, as per conditions specified in the labelling thereof and has not been approved as safe
and efficacious by the Central Licencing Authority with respect to its claims; or
(ii) A drug approved by the Central Licencing Authority for certain claims and proposed to be marketed
with modified or new claims including indication, route of administration, dosage and dosage form; or
(iii) A vaccine, recombinant Deoxyribonucleic Acid (r-DNA) derived product, living modified organism,
monoclonal anti-body, stem cell derived product, gene therapeutic product or xenografts, intended to be
used as drug;
As per second Schedule of New Drug and Clinical trial 2019, for new drug substances discovered or developed in countries other than India, Phase I data should be submitted along with the application. After submission of Phase I data generated outside India to the Central Licensing Authority, permission may be granted to repeat Phase I trials or to conduct Phase II trials and subsequently Phase III trial concurrently with other global trials for that drug. For a drug going to be introduced for the first time in the country, Phase III trial may be required to be conducted in India before permission to market the drug is granted unless otherwise exempted.
As per New Drugs and Clinical trial Rules 2019: the local clinical trial may not be required to be submitted along with the application if,
(i) The new drug is approved and marketed in countries specified by the Central Licencing Authority under rule 101 and if no major unexpected serious adverse events have been reported; or
(ii) The application is for import of a new drug for which the Central Licencing Authority had already granted permission to conduct a global clinical trial which is ongoing in India and in the meantime such new drug has been approved for marketing in a country specified under rule 101; and
(iii) There is no probability or evidence, on the basis of existing knowledge, of difference in Indian population of the enzymes or gene involved in the metabolism of the new drug or any factor affecting pharmacokinetics and pharmacodynamics, safety and efficacy of the new drug; and
(iv) The applicant has given an undertaking in writing to conduct Phase IV/PMS clinical trial to establish
further safety and effectiveness of such new drug as per design approved by the
•Provided that the Central Licencing Authority may relax this condition, where the drug is indicated in
life threatening or serious diseases or diseases of special relevance to Indian health scenario or for a condition
which is unmet need in India such as XDR tuberculosis, hepatitis C, H1N1, dengue, malaria, HIV, or for the
rare diseases for which drugs are not available or available at a high cost or if it is an orphan drug.
•As per Sixth Schedule: No fee shall be chargeable in respect of application for conduct of clinical trial for
orphan drugs as defined in clause (x) of rule 2.
As of now, CDSCO has granted approvals of imported and domestic Stem Cells and Stem cell derived products including avascular necrosis of hip joint, Articular Cartilage Defects (Autologous Nature), Buerger’s Disease with Critical Limb Ischemia, Scaffolding for correcting, reconstructing, filling, repairing or rebuilding bone in dental defects, etc.
• Reagents used for the derivation of human ESCs/iPSCs or expansion/enrichment of SSCs, for doing
Clinical Trials should be of Clinical Grade/pharmacopoeia grade.
• When using Research Grade material, the Quality Control Program” should check out for the safety,
efficacy and potency of the same as depicted in Annexure V, of NGCSR 2017.
• Animal-derived materials/reagents, for instance FCS (Fetal Calf Serum), Bovine Serum Albumin and
Trypsin/ Collagenase should be tested for ancillary agents (virus causing Spongiform Encephalopathy/
BSE).
• For all reagents imported from foreign countries, the country of distribution is to be clearly stated. The
FCS should be imported from BSE free country.
• Investigators should be encouraged to use Serum Free or Xeno free medium for processing and culturing
of stem cells.
Participants for the specified clinical trials should be selected as per the agreed “EXCLUSION” and
“INCLUSION” criteria of the designed protocol.
• Amendments/corrections/deviations should be immediately informed to CDSCO, IC-SCR and IEC.
• Human Participants enrolled for clinical trials are not liable to pay for any changes towards procedures.
Investigations, biochemical tests, and or hospitalization related to the trial.
• The “patient information sheet” and the “Informed Consent” form should be approved by IEC and ICSCR.
• The ongoing or current scenario in Stem Cell Research should categorically stated like, the duration,
investigational parameters of the proposed area of research, and possible “pros and cons” of the short term
or long term use of the same.
• Irreversibility of the investigation.
• The origin and salient features of the stem cells and the degree of Ex vivo manipulation, if done.
• The already substantiated standard of care available at the time of initiation, their merits and demerits.
• The sample size, dose, duration of study and follow ups.
• Copies of the IC-SCR and IEC approvals.
• The category of the clinical trial, that is whether they are randomized, double blinded, or open-labeled/
pilot studies.
• The participants of the clinical trial should be provided a copy of the “patient information sheet” and
“informed consent” immediately at the start and never deferred at a later date in local vernacular or language
for clear understanding of the motive behind such a trial.
• Video recording of the patient who is the participant & vulnerable in such a CT, according to the recent
amendment in CDSCO rule, viding, schedule Y, dated 9th January 2014.
• All clinical trials using Stem Cells and Stem Cell derived Products have to be registered with CTRI
(Available at http://ctri.nic.in/Clinicaltials/login.php).
• Only those institutions, which have their IC-SCR and IEC registered with CDSCO and NAC-SCRT
respectively are allowed to perform the clinical trials.
• CTs taking use of only minimally manipulated autologous Somatic Stem Cells (SSCs) that is HSCs and
MSCs, for homologous use for indications, other than those categorized in Annexure III of NGSCR 2017
or for non-homologous use, should have proper approval from NAC-SCRT, IC-SCR, CDSCO and IEC.
• CTs using stem cells manipulated substantially should have prior approval of IC-SCR, IEC and CDSCO.
• CTs making use of “ALLOGENIC” SSCs or Somatic Stem Cells, with any degree of manipulation, and
those using autologous SSCs, with more than minimal and major manipulation, should be approved by
CDSCO, IC-SCR and IEC.
• Any SCDP, being approved and marketed abroad or for concurrent CTs in Indian Hospitals, would require
approval of CDSCO, post clearance from IC-SCR and IEC.
• Any CTs, with a PRODUCT intended to be licensed and marketed in our country, should have prior
approval from IC-SCR and IEC.
• For tissue-engineered and combinatorial products, each of the components that are been undertaken in
CTs should be approved by CDSCO after being passed on from the rule book of IEC and IC-SCR.
A separate Data Safety Monitoring Board (DSMB), should be established before each and every clinical trial.
• All cases of “ADVERSE EFFECTS” or “SERIOUS ADVERSE EFFECTS” (AE/SAE) should be
immediately reported to CDSCO and IEC by the investigator/ researcher/Doctor/institution as clearly
mentioned in New Drugs and Clinical Trials Rules, 2019, of DRUG and COSMETICS act, 1940 or rules
established therein.
• The same should be reported to NAC-SCRT immediately via IC-SCR.
• Members of the DSMB are expected to be possessed with the required expertise and excellence to monitor
trials for AEs/SAEs and their smooth or trouble-free conduct.
• Members, being a part of DSMB, shall not have a semblance of Conflict of Interest with IC-SCR/
CDSCO/IEC.
• The institution/sponsor of the respective CTs, should provide the cover insurance to the participants and
compensation to the subjects enrolled for the same under trial.
• The Medical Record of the “Trial Participants” should be maintained for the period of at least fifteen (15)
years by head/CEOs of institutes via investigators and IC-SCR.
• Follow-up of the study participants is a must depending on the nature of the study protocol.
• Long term follow-up opens up the avenue for any late unwanted adverse effects and or fallacies in the
efficacies of the interventions.
• For each indication a minimum follow up of 2-3 years, is mandatory with respect to the safety data.
• The same should be approved and decided by the IC-SCR and IEC, on a case by case basis.
• Physical examination of the participants of each and every CTS is mandatory.
• The investigator should submit periodic report on follow up to the DSMB.
• Secretome is the sum total of different secretory molecules secreted by cells under the influence of inducers
or stimulators in “in vitro” or “ex vivo” studies done in relation to a particular pathological state. It is the
“conditioned medium”/CM that reflects the composition of the prospective “Secretome”.
• Current research involving proteomic profile linked with “FUNCTIONAL POTENCY of the Stem
Cells or Stem Cell-derived Products present within the conditioned medium or CM from Mesenchymal
Stem Cell Culture or of adipocytes, Bone Marrow, Wharton’s Jelly and the like.
• The CM has expressed the prospective usage of the so called Secretome in developing Extra-cellular
Matrix, Growth Factors, Chemokines and small molecules like peptides.
• For perusal as “active ingredient” for “COSMETIC” as well as for “TOPICAL” application, data on the
following should be submitted to CDSCO.
• Physical, chemical and pharmaceutical properties of the conditioned medium.
• Quantification of human cytokines or growth factors present in the conditioned medium.
• Composition of the final product and indications for its use in clinical trials.
• Pre-clinical studies for “TOXICITY” and “ALLERGENICITY”.
1. Safety studies and efficacy proofs of CM-PIPT (Primary Irritation Patch Test).
2. Human Volunteer study.
3. Based on the data that is submitted, allowance or approval for the responsible use of the conditioned
medium, may be granted as an active constituent for cosmetic or topical use on a case to case basis.
4. For use of CM as an active ingredient for interventional purposes, guidelines for product development as
provided in New Drugs & Clinical Trial Rules, 2019, of Drugs and Cosmetics Act 1940 or Rules present
therein.
The salient features of the National Guidelines for Stem Cell Research. 2017, in India is by no means a finished product. Stem Cell Science as a subject is still in its infancy and we are not by any stretch of imagination masters of this juvenile science. The translational potential of such an important pillar of modern medicine is noteworthy. We know very little about the mysteries of stem cell and its pre-clinical applications, let alone clinical applications. With time the virgin field of stem cell science and regenerative medicine, has taken rapid strides in recent times, so as a Science it is young and dynamic and so are its many ramifications.
It will be certainly beneficial to all concerned with the development of Stem Cell Science and Regenerative Medicine to inculcate new developments along its path of evolution and occasionally review and make amendments in the guidelines as per the advancement in knowledge. Accordingly periodic modification to specific clauses and sections, in the draft has to be enforced too. This is especially taken care by the Indian Council of Medical Research and they are given the responsibility to do the same and bring necessary changes as and when it is needed. Flexibility is the order of the day rather than getting too rigid about rules and guidelines.
Conclusions
Presently we are at the crossroads where drawbacks of common mode of therapies for different curable and
incurable diseases are well known and still not have the belief system ready to embrace the pillar of Modern
Medicine, Medical Devices and the “Realm of Stem Cell/Regenerative Medicine and Translational Sciences”.
BRegulatory approaches are taken and well categorized here in this review to highlight the importance of
the guidelines issued by NG-SCR, NAC-SCRT,CDSCO, ICSSR, ICMR, FDA, IEC etc., to minimize
mis-happenings and be a watchdog to curb unscrupulous individuals form undertaking unregulated, and
un-informed stem cell therapies to vulnerable and over-anxious patients.
While on the other hand many companies in India and abroad are making it a point to be aware and conversant with all the tenets of the issued guidelines and religiously implement them in their practice of procuring Stem Cells from different sources (autologous and allogenic, homologous or non-homologous) and Stem Cell derived Product applications. They adhere to every norms and mandates of different regulatory bodies and scrupulously brand and market their products. With the clear differentiation between research, regulation and therapies, industry, medical healthcare professionals and regulators will be able to provide their services for providing quality health standards in India. A clear definition of Stem cell and Stem cell derived products with xenografts needs to be provided by regulators for avoiding the puzzlement. Further, national laboratory with reference standards for regulatory strengthening in terms of Quality testing of Stem Cells and Stem Cell derived products is required to be setup in India as presently there is no notified laboratory for testing of Stem Cells and Stem Cell derived products.
Bibliography


Hi!
We're here to answer your questions!
Send us a message via Whatsapp, and we'll reply the moment we're available!